Alport sendromunun nedeni, böbreklerin, iç kulağın ve gözlerin düzgün çalışması için gerekli olan bir proteinin üretiminde yer alan genlerin mutasyonudur.
Alport sendromunun teşhisi fizik muayene, tıbbi öykü, böbrek biyopsisi ve genetik teste dayanır.
Şu anda Alport sendromundan muzdarip olanlar sadece semptomatik tedavilere güvenebilir, yani semptomları hafifletir ve komplikasyonları erteler.
Alport sendromu, "kalıtsal" teriminin "bir veya iki ebeveynden geçen" anlamına geldiği kalıtsal bir durumdur.
Epidemiyoloji: Alport Sendromu Ne Kadar Yaygındır?
İstatistiklere göre her 50.000 kişiden biri Alport sendromu ile doğuyor.
Alport Sendromunun Eş Anlamlıları
Kalıtsal doğası ve böbrek tutulumu nedeniyle Alport sendromu tıbbi olarak kalıtsal nefrit olarak da bilinir.
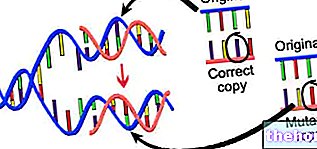
insanlar, hücre büyümesi ve replikasyonu da dahil olmak üzere yaşam için gerekli olan biyolojik süreçlerde temel proteinleri üretme görevine sahip DNA dizileridir.Mutasyonsuz olduklarında (dolayısıyla sağlıklı bir insanda), Alport sendromuyla ilişkili 3 gen, esas olarak böbreklerde bulunan tip IV kollajenin doğru üretimi, işlenmesi ve nihai yapılandırılması için temel olan bir protein bileşeni üretir. iç kulak ve gözlerde.
Öte yandan, mutasyon kurbanı olduklarında, Alport sendromu ile ilgili 3 gen, yukarıda bahsedilen tip IV kollajenin gerçekleşmesi için gerekli olan protein bileşenini üretme yeteneğini kaybeder ve bu, "böbreklerde, iç kulakta doku değişikliğini" içerir. ve gözler, sonuç olarak bu organların işlev bozuklukları.
Bunu biliyor muydun ...
Tip IV kollajenin Alport sendromu tarafından tutulumu, ikincisini Ehlers Danlos sendromu veya Marfan sendromu gibi bir bağ dokusu hastalığı yapar.
Alport sendromunun patofizyolojisi: biraz daha detay
Alport sendromuyla ilgili genler, aşağıdakiler için gerekli olan tip IV kollajen varyantının oluşumuna katkıda bulunur:
- Renal glomerüllerin kana karşı doğru filtreleme eylemi.
Açıklama. Böbreklerde bulunan renal glomerüller, kandaki atık maddeleri (sanki filtrelermiş gibi) çıkarabilen ve idrar oluşturabilen özel kan damarları aglomerasyonlarıdır.
Tip IV kolajenlerinin değişmesiyle bu böbrek yapıları filtreleme ve idrar oluşturma eylemlerinde başarısız olur.

- İç kulağın Corti organı tarafından ses dalgalarının sinir uyarılarına çevrilmesi işi.
Açıklama. Corti organı, kulak tarafından algılanan ses dalgalarını insan beyni tarafından "okunabilir" ve "yorumlanabilir" sinir sinyallerine dönüştürmekten sorumlu iç kulağın yapısıdır.
Tip IV kollajenin Corti organıyla birleşmesi, sesin insan sinir sisteminin diline çevrilme sürecini etkiler. - Lensin doğru şeklinin, korneanın yeterli sertliğinin ve retinanın pigmentli epitelinin normal renginin korunması.
Açıklama. Merceğin şekli ve korneanın sertliği görme fonksiyonu ve oküler sağlık için esastır (diğer taraftan retinanın pigmentli epitelinin rengi bu bölgelerde önemli görünmemektedir).
Gözlerde değiştirilmiş tip IV kolajenin varlığı, merceğin şeklini, kornea sertliğini ve retina pigment epitelinin rengini değiştirir.
Tüm bu bilgiler, Alport sendromunun neden belirli bir semptomatoloji ile ilişkili olduğunu açıklamaktadır.
Alport Sendromunun Kalıtımı
Anlamak...
- Her insan geni, biri anne kaynaklı ve biri baba kaynaklı olmak üzere alel adı verilen iki kopya halinde bulunur.
- Kalıtsal bir hastalık, mutasyona neden olan genin tek bir kopyası yeterli olduğunda otozomal dominanttır.
- Kalıtsal bir hastalık, mutasyona neden olan genin her iki kopyasının da meydana gelmesi gerektiğinde otozomal resesiftir.
Alport sendromunun 3 farklı kalıtım modeli vardır.
En yaygın kalıtım modeline göre (vakaların %80'i), Alport sendromu, üzerinde bulunan COL4A5 geninin mutasyonuna bağlı olduğundan, X kromozomuna bağlı (hemofili veya renk körlüğü gibi) kalıtsal bir hastalıktır. kromozom cinsel X.
İkinci en yaygın kalıtım modeline göre (vakaların yaklaşık %10'u), Alport sendromu, varlığı COL4A3 genlerinden birinin ve COL4A4'ün her iki alelinde de spesifik bir mutasyon gerektirdiğinden, otozomal resesif kalıtsal bir hastalık gibi davranır. 2. kromozom üzerinde.
Son olarak, daha az yaygın olan kalıtım modeline göre (vakaların yaklaşık %5'i), Alport sendromu otozomal dominant kalıtsal bir hastalığın bir örneğini temsil eder, çünkü COL4A3 ve COL4A4 genlerinin alellerinden sadece birinde spesifik bir mutasyon onun için yeterlidir. tezahür..
Alport Sendromu X'e Bağlı
X'e bağlı Alport sendromunun en kötü etkileri erkeklerde görülür.
Bu fenomen, erkekleri ve kadınları karakterize eden cinsel kromozomal yapı ile ilgilidir; aslında, kadınlarda, ikisinden birinde bir mutasyon olması durumunda, birbirine yardımcı olan iki X kromozomu vardır (sağlıklı olan, mutasyona uğramış olanın eksikliklerini az çok etkili bir şekilde telafi eder); "insan"da ise, sadece bir X kromozomu vardır (diğeri bir Y cinsiyet kromozomudur), eğer mutasyonlara maruz kalırsa, aynı işlevlere sahip başka herhangi bir kromozomun desteğine güvenemez.
Otozomal Resesif Alport Sendromu
Otozomal çekinik davranış gösteren Alport sendromu, hem erkek hem de kadınlarda benzer belirti ve bulgulara neden olur.
Otozomal Dominant Alport Sendromu
Otozomal baskın davranışa sahip Alport sendromu, erkekleri ve kadınları aynı şiddette etkiler.
ve trakeo-bronşiyal ağaç ve ciddi vasküler problemler (aort diseksiyonu ve abdominal veya torasik aort anevrizması).Böbrek Fonksiyonunun Kaybı: Belirtileri
Öncül"İlerleyen böbrek fonksiyonu kaybı" ile doktorlar, böbreklerin ve iç yapılarının (örneğin böbrek glomerülleri) filtreleme ve idrar üretim kapasitelerinde kademeli bir düşüşe maruz kaldığını kastetmektedir.
Alport sendromu varlığında böbrek fonksiyon kaybı ile ilgili semptomlar esas olarak hematüri yani idrarda kan ve proteinüri yani idrarda protein bulunmasından oluşur.
Hematüri, Alport sendromlu kişilerde genç yaşta ortaya çıkma eğiliminde olması anlamında oldukça erken bir semptomdur; çoğu durumda tanınması ancak mikroskopla (mikroskopik hematüri) mümkündür.
Tersine, proteinüri daha sonraki bir semptomdur, yani Alport sendromu ileri bir aşamadayken ortaya çıkar.
İşitme yeteneklerinde düşüş: ayrıntılar

Alport sendromu işitsel düzeyde kısmi sağırlığa neden olur; Kesin olmak gerekirse, hastanın yüksek frekanslardaki sesleri (yüksek frekanslı işitme kaybı) duymasını engelleyen bir "sensörinöral işitme kaybına" neden olur.
Alport sendromundan muzdarip kişilerde işitme sorunlarının gelişimi, mutasyona maruz kalan gene ve kalıtımın türüne göre değişir; aslında:
- Mutasyon COL4A5'te bulunuyorsa, erkeklerde bebekliğin sonlarına doğru işitme kaybı gelişirken, kadınlarda, etkilendikleri nadir durumlarda daha sonraki yaşlarda gelişir;
- Mutasyon COL4A4 veya COL4A3'te bulunuyorsa ve hastalık otozomal resesif ise, hem erkek hasta hem de kadın hasta, işitme kaybının ilk belirtilerinin geç çocukluk veya erken ergenlik döneminden şikayet eder;
- Mutasyon COL4A4 veya COL4A3'te bulunuyorsa ve hastalık otozomal dominant ise, hem erkek hem de kadın hastalarda ileri yaşlarda sensörinöral işitme kaybı gelişir.
Hala bilinmeyen nedenlerle Alport sendromu bazı hastaları işitme problemlerinden kurtarır; başka bir deyişle, bilinmeyen nedenlerle Alport sendromlu bazı kişilerde herhangi bir sensörinöral işitme kaybı gelişmez.
Oküler Anomaliler ve Görme Eksiklikleri: ayrıntılar
Hastaların sadece belirli bir yüzdesinde gözlemlenebilen Alport sendromunun neden olduğu oküler anomaliler, keratokonus, lentikon, katarakt ve retina makulasında lekelerin varlığı gibi durumların nedenidir.
Retina makula üzerinde lekelerin varlığı dışında, yukarıda bahsedilen tüm bu durumlar, hastaların görme yetilerini aşağı yukarı derinden etkiler.
Özofagus ve Trakeo-Bronşiyal Ağacın Diffüz Leiomyomatozisi
Özofagusun ve trakeobronşiyal ağacın diffüz leiomyomatozisi, Alport sendromlu kişilerde aşağıdakilere neden olabilen nadir iyi huylu tümörlerdir:
- disfaji;
- tokluk kusma
- Epigastrik ağrı ve retrosternal ağrı;
- Tekrarlayan bronşit;
- dispne;
- Öksürük;
- Nefes alırken hıçkırık.
Alport Sendromu: Komplikasyonlar
Alport sendromunun ana komplikasyonu, böbrek fonksiyonunun ilerleyici ve amansız kaybından kaynaklanan son aşamada böbrek yetmezliği durumudur.
Tıpta "son aşama" böbrek yetmezliği terimi, kronik böbrek yetmezliğinin en şiddetli aşamasının yanı sıra son aşamayı, yani böbreklerin tüm fonksiyonel kapasitelerini tamamen ve kalıcı olarak kaybettiği durumu belirtir.
Çok ciddi sonuçlardan (örn: hipertansiyon, pulmoner ödem, kemik kırılganlığı, immünosupresyon, sinir sistemi hasarı vb.), "son aşama böbrek yetmezliği, diyaliz gibi kronik tedavi gerektirir ve" ameliyat ameliyatı için en önemli endikasyondur. örneğin böbrek nakli.
Diğer Komplikasyonlar
Alport sendromunun ölüm oranı yüksek olan diğer olası komplikasyonları, torasik veya abdominal aortun "olası aort diseksiyonu veya" olası anevrizmasının yırtılmasını takiben iç kanamadır.
böbrek;Fiziksel inceleme
Hastanın sergilediği belirti ve bulguların tıbbi olarak gözlemlenmesidir.
Alport sendromu bağlamında, böbrek problemlerinin, (varsa) işitme problemlerinin ve (varsa) oküler anormalliklerin bir kısmını ortaya çıkarır.
anamnez
Hastanın aile öyküsünün incelenmesiyle birlikte, belirli sorular aracılığıyla semptomların eleştirel olarak incelenmesidir.
Alport sendromunu tanımaya yarayan muayene sürecinde, anamnez, doktorun semptomların tam olarak başladığı anı ve hastanın ailesinde kalıtsal bir hastalık olup olmadığını bilmesini sağlar.
böbrek biyopsisi
Bir böbrek hücresi örneğinin toplanması ve laboratuvar analizinden oluşur.
Alport sendromu bağlamında, böbreklerin sağlık durumunu değerlendirmek ve fonksiyonel tip IV kollajenin yokluğunu tespit etmek yararlıdır.
Böbrek biyopsisi Alport sendromu tanısında önemli bir adımdır, çünkü zamanında yapılırsa en uygun tedavinin en kısa sürede planlanmasına olanak tanır.
genetik test
Kritik genlerdeki mutasyonları tespit etmeyi amaçlayan DNA analizidir.
Alport sendromu bağlamında, doğrulayıcı tanı testini ve kesin mutasyona uğramış genin ve kalıtım tipinin belirleneceği testi temsil eder.
ACE-İnhibitörleri
Normalde hipertansiyon ve bazı kardiyovasküler hastalıkların tedavisi için endike olan ACE inhibitörleri, yukarıda bahsedilen kalıtsal hastalığın tipik özelliği olan böbrek fonksiyonlarının ilerleyici düşüşünü yavaşlatma yetenekleri sayesinde Alport sendromunun varlığında kullanılır.
Dolayısıyla bu yetenek sayesinde ACE inhibitörleri, böbrek yetmezliğinin başlangıcını ve ayrıca diyaliz veya böbrek nakli gibi önemli tedavilere duyulan ihtiyacı ertelemeye izin verir.
Bilinmeyen nedenlerle, ACE inhibitörleri Alport sendromlu bazı kişilerde etkisizdir; bu, doktorların neden benzer güce sahip ilaçları aradıklarını açıklıyor.
Kronik Böbrek Yetmezliği için Farmakolojik Tedaviler
Alport sendromu kronik böbrek yetmezliği ile sonuçlandığında, aşağıdakiler de dahil olmak üzere birkaç yararlı ilaç vardır:
- Hipertansiyona karşı ilaçlar (bunlar arasında yukarıda bahsedilen ACE inhibitörleri ve ARB'ler ve diüretikler);
- Kalsiyum ve D vitamini takviyeleri (kemikleri kırıklardan korumak için);
- Sodyum polistiren sülfonat ve analogları (kanda potasyum birikmesini önlemek için).
Diyaliz ve Böbrek Nakli
Diyaliz, böbreğin belirli işlevlerini yapay olarak yeniden üreten, kanı fazla atık ürünlerden ve sudan temizleyen bir tedavidir.
Böbrek nakli ise böbreklerden birinin veya her ikisinin uyumlu bir donörden (verici cansız veya canlı olabilir) sağlıklı bir böbrekle değiştirilmesi ameliyatıdır.
Hem diyaliz hem de böbrek nakli, Alport sendromunun en ileri evrelerinde, yani böbrek yetmezliğinin son evresinde olduğu durumlarda endikedir.
İşitme cihazı
Alport sendromunun varlığında işitme cihazı, kısmi sağırlığı olan tüm hastalar için uygun bir çözümdür.
Keratokonus, Lentikon ve Katarakt Tedavisi
Alport sendromu gözleri etkileyerek keratokonus, lentikon ve/veya kataraktlara neden olduğunda, hastalar göz ameliyatına güvenebilir ve sonuçları tatmin edici olmaktan ötedir.
Alport Sendromunun Semptomatik Tedavisi hangi tıbbi figürleri içerir?
Alport sendromunun semptomatik tedavisi, çocuk doktorları, nefrologlar, odyologlar ve oftalmologlar dahil olmak üzere birkaç tıp uzmanının koordineli müdahalesini gerektirir.

-cos-cause-e-terapia.jpg)