Aniridia da öyle
Aniridia, irisin tamamen veya kısmen yokluğu ile karakterize doğuştan gelen bir hastalıktır. Bu durum kalıtsal, sporadik veya bir sendromun parçası olabilir.
Aniridia, irisin gelişimsel bir kusuru ile sınırlı değildir, ancak genellikle doğumdan veya geç başlangıçlı bir dizi oküler komplikasyonla ilişkilidir: azalmış görme keskinliği, maküler ve optik sinir hipoplazisi, nistagmus, ambliyopi, katarakt ve kornea değişiklikleri.

nedenler
Aniridia, PAX6 genini içeren kromozom 11'in (11p13) kısa kolunun AN2 bölgesini etkileyen genetik bir değişiklikten veya ekspresyonunu kontrol eden dizilerin silinmesinden kaynaklanır. PAX6, gözün ve beyin, omurilik ve pankreas gibi diğer oküler olmayan yapıların erken gelişiminde yer alan bir dizi başka olayı yöneten talimatlar sağlar.
Hastalığın klinik ifadesi değişkendir ve 11p13 bölgesini etkileyen değişikliğin tipine bağlıdır (mutasyon, delesyon, translokasyon, yerleştirme, vb.) Bazı kusurlar PAX6 protein ürünlerinin işlevini daha fazla engellerken, diğerleri ifadeyi garanti eder. hafif bir fenotip üretmek için yeterli gen.
İzole aniridi. Hastalık ilk kez taşıyıcı olmayan ebeveynlerden doğan bir çocukta kendini gösterebilir. Aniridinin başlamasına neden olan sporadik mutasyonlar, kromozom 11p13'teki de novo delesyonlar nedeniyle sistemik tutulum olmaksızın ortaya çıkabilir.
Sendromik aniridi. Az sayıda vakada, genetik kusur AN2 bölgesine bitişik WT1 bölgesini etkiliyorsa, hastalık bir sendrom içinde başlayabilir ve nefroblastom (Wilms tümörü) veya diğer kusurlar gibi diğer oküler olmayan anomalilerle ilişkili olabilir. .
Vizyon üzerindeki sonuçlar
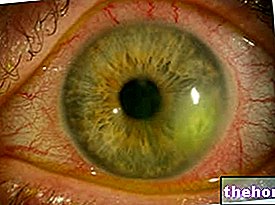
Aniridia, diğer az ya da çok ciddi oküler özelliklerle ilişkili olarak, her iki gözde irisin yokluğuna kadar değişen derecelerde hipoplazi (eksik gelişme) ile kendini gösterir:
- Çok büyük veya deforme olmuş öğrenci, ışığa ve parlamaya karşı aşırı duyarlılığı belirler.
- Korneanın vaskülaritesi ve opaklığı ışığın kırılmasında kusurlara neden olabilir.
- Katarakt (merceğin bulanıklaşması) hastaların %50-80'inde görülür ve görüşü bozabilir.
- Lens sublukse olabilir (normal konumundan kayabilir).
- Foveanın (maksimum görme keskinliğine sahip retinanın merkezi alanı) veya optik sinirin zayıf gelişimi, nistagmusa ve görme azalmasına neden olabilir.
- Aniridisi olan kişilerin %50'sinden fazlasında glokom gelişir (retina ve optik sinirde kalıcı hasara neden olabilen yüksek göz içi basıncı).
Diğer sağlık sorunları
Aniridia, diğer vücut sistemleri için herhangi bir ima olmaksızın veya bir sendromun (sürekli birlikte ortaya çıkan bir grup belirti) parçası olarak ortaya çıkabilir.
- Miller sendromunda, aniridia böbreğin malign neoplazmı ile ilişkilidir: Wilms' tümörü (nefroblastoma).
- Gillespie sendromu, aniridi, zeka geriliği ve denge problemlerinin (serebellar ataksi) bir kombinasyonudur.
- WAGR sendromu aniridi, genital anormallikler, zeka geriliği ve Wilms tümörü ile kendini gösterir.
Teşhis ve tedavi
Aniridinin ana tanısal özelliği, irisin kısmen veya tamamen yokluğudur; azalmış görme keskinliği ile fovea hipoplazisi hemen hemen her zaman mevcuttur ve nistagmusun erken başlangıcı ile ilişkilidir. Genellikle geç başlangıçlı diğer oküler anormallikler, görme fonksiyonunun daha da azalmasına neden olur ve dikkatle izlenmelidir. Bu koşullar şunları içerir: katarakt, glokom ve kornea opaklaşması. Aniridi teşhisi konduktan sonra, bu nedenle, genellikle yaşam boyunca periyodik göz muayeneleri gereklidir. Ziyaretlerin sıklığı, klinik ifadeye ve bozukluğun yaygınlığına bağlıdır. PAX6 gen delesyonu olan çocuklarda. Böbrek fonksiyonunun izlenmesi ve sık ultrason kontrolleri önerilir. .
Terapötik yaklaşım, görme keskinliğini iyileştirmeyi ve aniridi ile ilişkili herhangi bir komplikasyonu farmakolojik veya cerrahi olarak yönetmeyi amaçlar.Gözlük reçetesi kırma kusurlarını düzeltebilir.Reçeteli renkli veya fotokromik lenslerin kullanılması, bunun yerine parlamayı önlemek ve ışığa karşı aşırı duyarlılığı azaltmak için yararlıdır.











-caroteni-misti-e160-a-(ii)--carotene.jpg)





-cos-sintomi-e-primo-soccorso.jpg)