genellik
Dönem retinitis pigmentoza (RP), ilerleyici retina dejenerasyonu ile karakterize edilen bir grup genetik hastalığı tanımlar.

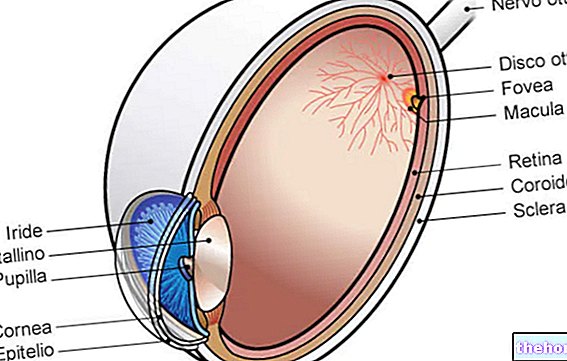
Retinitis pigmentosa, kademeli fotoreseptör kaybı ve pigment epitelinin işlev bozukluğu ile karakterize bir retina distrofisidir.Bu, retinanın optik sinir yoluyla beyne görsel bilgi iletme yeteneğini giderek azalttığı anlamına gelir.
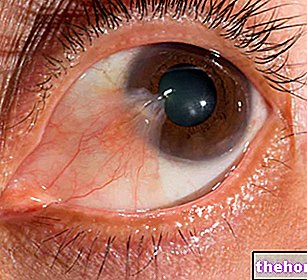
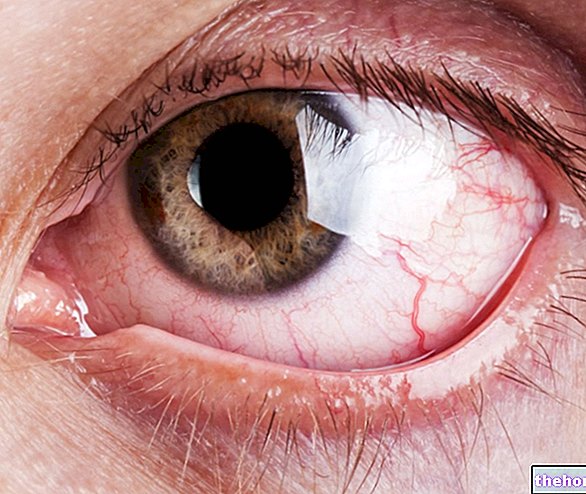
Patolojik süreç, retina pigment epitelindeki değişikliklerle başlar.Retinitis pigmentosa ilerledikçe, retinayı besleyen kan damarlarında incelme olur ve atrofiye uğrar.Fundus muayenesi üzerine karakteristik tortular görsel olarak saptanabilir. dolayısıyla hastalığın adı). Atrofik değişiklikler ve hasar ayrıca optik siniri de içerebilir ve yavaş yavaş retinanın ışığa duyarlı hücreleri ölür.
Retinitis pigmentosa'dan etkilenen hastalar, özellikle yetersiz aydınlatılmış ortamlarda başlangıçta görme sorunları yaşarlar ve periferik görme alanında daralmadan şikayet ederler. Merkezi görme, hastalığın sonraki aşamalarına kadar korunur ve nihai sonuç önemli ölçüde değişebilir: retinitis pigmentosa'lı birçok insan, yaşamları boyunca sınırlı bir görüşe sahip olurken, diğerleri tamamen görme yeteneğini kaybeder.
Retinitis pigmentosa, esas olarak bir veya iki ebeveynden geçen genetik değişikliklerin neden olduğu kalıtsal bir hastalıktır. Genetik kusurun türü, bozukluğa en çok hangi retina hücrelerinin dahil olduğunu belirler ve klinik açıdan farklı koşulları ayırt etmeyi mümkün kılar. Bugüne kadar, retinitis pigmentosa ile ilişkili 50'den fazla farklı genetik kusur tanımlanmıştır. Anormallikler, üç kalıtım modelinden biri yoluyla ebeveynlerden yavrulara geçebilir: otozomal çekinik, otozomal dominant veya heterozomal çekinik (X'e bağlı veya X'e bağlı).
Belirtiler
Daha fazla bilgi için: Retinitis Pigmentosa Belirtileri
Retinitis pigmentosa genellikle ergenlerde ve genç erişkinlerde bulunur. Semptomlar genellikle 10 ila 30 yaşları arasında ortaya çıkar, ancak tanı erken çocuklukta veya yaşamın çok daha ileri dönemlerinde yapılabilir.
Retinitis pigmentosa'nın erken belirtileri şunları içerebilir:
- Geceleri görme zorluğu (gece körlüğü) veya düşük ışık koşullarında
- Karanlıkta görüşten ışıkta görüşe yavaş adaptasyon ve tam tersi;
- Görme alanının daralması ve çevresel görme kaybı;
- Işığa ve parlamaya karşı hassasiyet.
Bazı semptomlar, ilgili fotoreseptörlerin tipine bağlıdır. Çubuklar siyah beyaz görmeden sorumludur, koniler ise renkleri ayırt etmenizi sağlar.
Çoğu retinitis pigmentosa vakasında, önce çubuklar etkilenir. Bununla birlikte, hızla gelişen formlarda, koniler de erken bir aşamada etkilenebilir.
Çubuklar retinanın dış kısımlarında yoğunlaşır ve loş ışıkla aktive olur, bu nedenle dejenerasyonları periferik ve gece görüşünü etkiler. Koniler dahil ise, renk algısı ve merkezi görme kaybı yaşanabilir.
İlgili fotoreseptörlerin baskınlığı, hastanın genetik yapısında mevcut olan belirli kusur tarafından belirlenir.
Çoğu zaman, retinitis pigmentosa'nın ilk belirtisi gece körlüğüdür (veya noktalopi). Bazı insanlar, iyi aydınlatılmış bir alandan daha karanlık bir alana geçerken ışıktaki farklılıklara uyum sağlamak için daha fazla zamana ihtiyaçları olduğunu fark eder. Tipik bir görme kaybı şekli, çevresel görüşün daralmasına neden olur (tünel veya teleskop görüşü); bu desene halka skotom denir. Bazen bu fenomen erken evrelerde gözden kaçabilir, ancak kişi sıklıkla nesnelerin üzerinden geçtiğinde veya bir trafik kazası geçirdiğinde fark edilir.Görme kaybı retinanın orta bölgesini kapsadığında (maküler distrofi olarak da adlandırılır) hastalar İpliği iğne deliğinden geçirmek gibi tek bir nesne üzerinde konsantrasyon gerektiren okuma ve ayrıntılı işlerde zorluk yaşar Birçok hasta, genellikle küçük, titreyen ve parıldayan ışıklar olarak tanımlanan ışık parlamaları (fotopsi) gördüğünü bildirir.
Hastalığın ilerleme hızı ve görme kaybının derecesi kişiden kişiye değişir. Bazı uç vakalar yirmi yıl içinde hızla gelişebilirken, diğerleri asla tam körlüğe yol açmayan yavaş bir seyir izleyebilir. Erken başlangıç, daha şiddetli retinitis pigmentosa formlarında bulunurken, daha hafif koşullara (örneğin otozomal dominant) sahip hastalar, hastalığı yaşamlarının beşinci veya altıncı on yılında geliştirebilir.X'e bağlı retinitis pigmentosa olan ailelerde, erkekler daha sık etkilenir Kadınlara göre ve daha şiddetli olarak, dişiler ise genetik özelliği iletir (değiştirilmiş geni X kromozomunda taşırlar) ve bozukluğun semptomlarını daha az sıklıkla gösterirler.
komplikasyonlar
Retinitis pigmentosa yavaş da olsa ilerlemeye devam edecektir. Bununla birlikte, tam körlük nadirdir, ancak periferik ve merkezi görmede önemli azalma meydana gelebilir.
Retinitis pigmentosa olan hastalarda genellikle erken yaşta retinada şişme (maküler ödem) veya katarakt gelişir. Bu komplikasyonlar görüşe müdahale ederse tedavi edilebilir.
İlgili hastalıklar
Genellikle, retinitis pigmentosa olan bir hastada başka bir bozukluk yoktur ve bu durumda "non-sendromik" veya basit retinitis pigmentosa'dan bahsediyoruz. Bununla birlikte, bazı sendromlar bu göz hastalığıyla bazı klinik semptomları paylaşır; en yaygın olanı, retinitis pigmentosa'lı tüm hastaların yaklaşık %10-30'unu etkileyen ve eş zamanlı konjenital veya ilerleyici işitme kaybı ile ilişkili olan Usher sendromudur. Bununla birlikte, Leber'in konjenital amorozunda, çocuklar yaşamın ilk altı ayında kör olabilir veya neredeyse kör olabilir.Retinitis pigmentosa ile ilgili diğer hastalıklar arasında Bardet-Biedl sendromu ve Refsum hastalığı bulunur.
nedenler
Hastalığa bir dizi genetik kusur neden olabilir: aslında, değişiklikten etkilenirse retinitis pigmentosa fenotipine neden olabilen birkaç gen vardır.Bunlar normal olarak, görmeyi sağlayan transdüksiyon kaskadı ile ilgili proteinleri kodlar, hücre transkripsiyonunu etkiler (retinal hücrelere hatalı mesajlar gönderirler) veya fotoreseptörlerin yapısını oluşturan elementler için Kalıtsal gen mutasyonları hücrelerde döllenme anından itibaren mevcuttur; yaygın anormallikler RP1 genlerininkileri içerir (retinitis pigmentosa-1, otozomal dominant) , RHO (RP4, otozomal dominant) ve RDS (RP7, otozomal dominant) Retinitis pigmentosa'nın kalıtsal olmayan nedenleri nadirdir, ancak aile öyküsü olmayan izole bir vaka (spontan mutasyon) bulma olasılığı. hastalık.