Aktif maddeler: Sorafenib
Nexavar 200 mg film kaplı tabletler
Nexavar neden kullanılır? Bu ne için?
Nexavar hepatokarsinom tedavisinde kullanılır.
Nexavar ayrıca böbrek kanseri (ileri böbrek hücreli karsinom) ileri bir aşamada olduğunda ve standart tedavinin onu durdurmaya yardımcı olmadığı veya uygun olmadığı düşünüldüğünde tedavi etmek için kullanılır.
Nexavar, tiroid kanserini (farklılaşmış tiroid kanseri) tedavi etmek için kullanılır.
Nexavar, sözde bir çoklu kinaz inhibitörüdür. Kanser hücrelerinin büyüme hızını yavaşlatarak ve kanser hücrelerinin büyümesine izin veren kan akışını bloke ederek çalışır.
Kontrendikasyonlar Nexavar ne zaman kullanılmamalıdır?
Nexavar'ı kullanmayınız.
- Sorafenib veya bu ilacın içerdiği diğer maddelerden herhangi birine karşı alerjiniz varsa (bölüm 6'da listelenmiştir).
Kullanım Önlemleri Nexavar'ı almadan önce bilmeniz gerekenler
Nexavar'ı almadan önce doktorunuz veya eczacınız ile konuşunuz.
Özellikle Nexavar'a özellikle dikkat edin
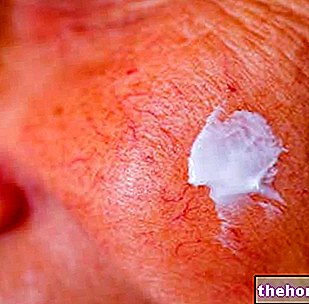
- Cilt sorunları ortaya çıkarsa. Nexavar, özellikle eller ve ayaklarda kızarıklıklara ve cilt reaksiyonlarına neden olabilir. Bu etkiler genellikle doktor tarafından tedavi edilebilir. Aksi takdirde doktor tedaviyi askıya alabilir veya tamamen durdurabilir.
- Yüksek tansiyonunuz varsa. Nexavar kan basıncında artışa neden olabilir; doktorunuz kan basıncınızı düzenli olarak kontrol edecek ve yüksek tansiyon tedavisi için ilaçlar reçete edebilir.
- Kanama probleminiz varsa veya varfarin veya fenprokomon alıyorsanız. Nexavar ile tedavi kanama riskinde artışa neden olabilir. Kan pıhtılarını önlemek için kanı incelten ilaçlar olan varfarin veya fenprokomon kullanıyorsanız, kanama riskinde artış olabilir.
- Göğüs ağrınız veya kalp probleminiz varsa. Doktorunuz tedaviyi durdurmaya veya tamamen durdurmaya karar verebilir.
- 'QT uzaması' olarak adlandırılan elektrik sinyali bozukluğu gibi bir kalp rahatsızlığınız varsa.
- Ameliyat olmak üzereyseniz veya yeni geçirdiyseniz. Nexavar yara iyileşmesini etkileyebilir. Ameliyat olmak üzereyseniz, Nexavar ile tedaviniz muhtemelen durdurulacaktır. Doktorunuz daha sonra ne zaman geri alacağınıza karar verecektir.
- Aynı zamanda kanser ilaçları olan irinotekan veya docetaxel ile tedavi ediliyorsanız, Nexavar bu ilaçların etkilerini ve özellikle yan etkilerini artırabilir.
- Neomisin veya başka antibiyotik alıyorsanız. Nexavar'ın etkinliği azalabilir - Şiddetli karaciğer yetmezliğiniz varsa Bu ilacı kullanırken yan etkileriniz kötüleşebilir.
- Böbrek fonksiyonunuz azalmışsa. Doktorunuz su ve elektrolit dengenizi izleyecektir.
- Doğurganlık. Nexavar hem erkeklerde hem de kadınlarda doğurganlığı azaltabilir. Bu sizin için geçerliyse, doktorunuzla konuşun.
- Tedavi sırasında gastrointestinal perforasyon meydana gelebilir (bkz. bölüm 4: Olası yan etkiler). Bu durumda doktor tedaviyi durduracaktır.
- Tiroid kanseriniz varsa, doktorunuz kanınızdaki kalsiyum ve tiroid hormonu düzeylerini kontrol edecektir.
Bunlardan herhangi biri sizin için geçerliyse doktorunuza söyleyiniz. Bu sorunlar için tedaviye ihtiyacınız olabilir veya doktorunuz Nexavar dozunu değiştirebilir veya tedaviyi tamamen durdurabilir (ayrıca bkz. bölüm 4: Olası yan etkiler).
Çocuklar ve ergenler
Nexavar henüz çocuklarda ve ergenlerde çalışılmamıştır.
Etkileşimler Hangi ilaçlar veya yiyecekler Nexavar'ın etkisini değiştirebilir
Bazı ilaçlar Nexavar'ı etkileyebilir veya ondan etkilenebilir. Bu listedeki ilaçlardan herhangi birini veya reçetesiz alınanlar da dahil olmak üzere diğer ilaçları alıyorsanız, yakın zamanda aldıysanız veya alma ihtimaliniz varsa doktorunuza veya eczacınıza bunlar hakkında bilgi veriniz:
- Rifampisin, neomisin veya enfeksiyonları tedavi etmek için kullanılan diğer ilaçlar (antibiyotikler)
- John's Wort olarak da bilinen Hypericum perforatum, depresyon için bitkisel bir tedavi
- Fenitoin, karbamazepin veya fenobarbital, epilepsi ve diğer hastalıklar için tedaviler
- Çeşitli hastalıklar için kullanılan bir kortikosteroid olan deksametazon
- Varfarin veya fenprokomon, kan pıhtılarını önlemek için kullanılan antikoagülanlar
- Kanser tedavisinde kullanılan doksorubisin, kapesitabin, dosetaksel, paklitaksel ve irinotekan.
- Hafif veya orta dereceli kalp yetmezliğinin tedavisinde kullanılan digoksin
Uyarılar Şunları bilmek önemlidir:
Hamilelik ve emzirme
Nexavar ile tedavi edilirken hamile kalmaktan kaçının. Çocuk doğurma çağındaysanız, Nexavar tedavisi sırasında etkili doğum kontrolü kullanmalısınız. Nexavar tedavisi görürken hamile kalırsanız, tedaviye devam edilip edilmeyeceğine kimin karar vereceğini derhal doktorunuza söyleyiniz.
Nexavar tedavisi görürken bebeğinizi emzirmemelisiniz, çünkü bu ilaç bebeğinizin büyümesini ve gelişmesini etkileyebilir.
Araç ve makine kullanma
Nexavar'ın araç veya makine kullanma yeteneğini etkileyeceğine inanmak için hiçbir neden yoktur.
Doz, Yöntem ve Uygulama Süresi Nexavar nasıl kullanılır: Pozoloji
Yetişkinler için önerilen Nexavar dozu günde iki kez iki adet 200 mg tablettir.
Bunlar günlük 800 mg doza veya günde dört tablete karşılık gelir. Nexavar tabletleri bir bardak su ile birlikte, öğün aralarında veya düşük ila orta yağlı yiyeceklerle birlikte alınız.Bu ilacı çok yağlı yiyeceklerle birlikte almayınız, çünkü bunlar etkilerini azaltabilir. Çok yağlı yiyecekler yemeyi planlıyorsanız, tabletleri öğle yemeğinden en az 1 saat önce veya 2 saat sonra alınız. Bu ilacı her zaman tam olarak doktorunuzun size söylediği şekilde alınız. Şüpheniz varsa, doktorunuza veya eczacınıza danışın.
Kandaki konsantrasyonu sabit tutmak için bu ilacı her gün yaklaşık aynı saatte almak önemlidir.
Bu ilaç genellikle klinik fayda sağlandığı sürece alınır ve hiçbir dayanılmaz yan etkisi yoktur.
Aşırı doz Çok fazla Nexavar aldıysanız ne yapmalısınız?
Kullanmanız gerekenden daha fazla Nexavar kullandıysanız
Siz veya bir başkası reçete edilen dozdan fazlasını aldıysa derhal doktorunuza söyleyiniz. Çok fazla Nexavar almak, özellikle ishal ve cilt reaksiyonları olmak üzere yan etkileri daha olası veya daha ciddi hale getirir. Doktorunuz size bu ilacı almayı bırakmanızı söyleyebilir.
Nexavar'ı kullanmayı unutursanız
Bir doz almayı unuttuysanız, hatırladığınız anda onu alınız. Bir sonraki dozunuzun zamanı azsa, unuttuğunuz dozu unutun ve sık kullandığınız dozla devam edin.
Yan Etkiler Nexavar'ın yan etkileri nelerdir?
Tüm ilaçlar gibi, bu ilaç da yan etkilere neden olabilir, ancak bunları herkes almayabilir. Bu ilaç ayrıca bazı kan testlerinin sonuçlarını da değiştirebilir.
Çok yaygın:
10 kişiden 1'inden fazlasını etkileyebilir
- ishal
- halsizlik (mide bulantısı)
- zayıf veya yorgun hissetmek (yorgunluk)
- ağrı (ağız ağrısı, karın, baş ağrısı, kemik ağrısı, kanser ağrısı dahil)
- saç dökülmesi (alopesi)
- avuç içlerinde veya ayak tabanlarında kızarıklık veya ağrı (el-ayak deri reaksiyonu)
- kaşıntı veya kızarıklık
- öğürdü
- kanama (beyinde, bağırsak duvarında ve solunum yollarında kanama dahil)
- yüksek tansiyon veya artan kan basıncı (hipertansiyon)
- enfeksiyonlar
- iştahsızlık (anoreksi)
- kabızlık
- eklem ağrısı (artralji)
- ateş
- kilo kaybı
- cilt kuruluğu
Yaygın:
10 kişiden 1'ini etkileyebilir
- grip benzeri hastalık
- hazımsızlık (dispepsi)
- yutma güçlüğü (disfaji)
- ağız iltihabı veya kuruluğu, dilde ağrı (stomatit ve mukoza zarı iltihabı)
- kanda düşük kalsiyum seviyeleri (hipokalsemi)
- kanda düşük potasyum seviyeleri (hipokalemi)
- kas ağrısı (miyalji)
- Karıncalanma ve uyuşma dahil el ve ayak parmaklarında hassasiyet bozuklukları (periferik duyusal nöropati)
- depresyon
- ereksiyon sorunları (iktidarsızlık)
- ses değişiklikleri (disfoni)
- akne
- iltihaplı, kuru veya pullanan cilt (dermatit, cilt soyulması)
- kalp yetmezliği
- kalp krizi (miyokard enfarktüsü) veya göğüs ağrısı
- kulak çınlaması (kulaklarda çınlama)
- böbrek yetmezliği
- idrarda yüksek protein seviyeleri (proteinüri)
- genel zayıflık veya güç kaybı (asteni)
- Beyaz kan hücrelerinin sayısında azalma (lökopeni ve nötropeni)
- kırmızı kan hücrelerinin sayısında azalma (anemi)
- kanda düşük trombosit sayısı (trombositopeni)
- saç köklerinin iltihabı (folikülit)
- azalmış tiroid aktivitesi (hipotiroidizm)
- kanda düşük sodyum seviyeleri (hiponatremi)
- tat alma duyusunda değişiklikler (disguzi)
- yüzde ve sıklıkla cildin diğer bölgelerinde kızarıklık (kızarma)
- burun akıntısı (burun akıntısı)
- mide ekşimesi (gastroözofageal reflü hastalığı)
- cilt kanseri (keratoakantom / skuamöz hücreli cilt kanseri)
- derinin dış tabakasının kalınlaşması (hiperkeratoz)
- bir kasın ani istemsiz kasılması (kas spazmları)
Yaygın olmayan:
100 kişiden 1'ini etkileyebilir
- mide iltihabı (gastrit)
- Pankreatite bağlı mide (karın) ağrısı, safra kesesi ve/veya safra yollarında iltihaplanma
- Safra pigmentlerinin yüksek seviyelerinin (hiperbilirubinemi) neden olduğu ciltte veya gözlerde sararma (sarılık)
- alerjik tip reaksiyonlar (cilt reaksiyonları ve kurdeşen dahil)
- dehidrasyon
- meme büyütme (jinekomasti)
- nefes almada zorluk (akciğer hastalığı)
- egzama
- aşırı tiroid aktivitesi (hipertiroidizm)
- çoklu deri döküntüleri (eritema multiforme)
- yüksek kan basıncı
- gastrointestinal perforasyon
- Baş ağrısı, bilinç değişikliği, nöbetler ve görme kaybı dahil görsel semptomlar (arka geri dönüşümlü lökoensefalopati) ile ilişkili olabilen beynin arkasında geri dönüşümlü ödem
- ani, şiddetli alerjik reaksiyon (anafilaktik reaksiyon)
Nadir:
1000 kişiden 1'ini etkileyebilir
- Solunum ve yutma güçlüğüne (anjiyoödem) neden olabilen derinin şişmesi (örn. yüz, dil) ile alerjik reaksiyon
- anormal kalp ritmi (QT uzaması)
- Mide bulantısı, kusma, karın ağrısı ve sarılığa (ilaca bağlı hepatit) yol açabilen karaciğer iltihabı
- Daha önce radyasyon tedavisine maruz kalmış ve şiddetli olabilen (aktinik benzeri dermatit) ciltte güneş yanığı benzeri döküntü
- Geniş cilt alanlarının ayrılmasıyla birlikte ağrılı kabarcıklar ve ateşi içerebilen ciddi cilt ve/veya mukoza zarları reaksiyonları (Stevens-Johnson sendromu ve toksik epidermal nekroliz)
- Böbrek problemlerine yol açabilen anormal kas hasarı (rabdomiyoliz)
- idrarda büyük miktarda protein kaybına neden olan böbrek hasarı (nefrotik sendrom)
- Derideki kan damarlarının döküntü şeklinde kendini gösterebilen iltihabı (lökositoklastik vaskülit)
Bilinmeyen:
mevcut verilerden sıklık tahmin edilemez
- örneğin uyku hali, davranış değişiklikleri veya kafa karışıklığı (ensefalopati) ile ilişkili olabilen bozulmuş beyin fonksiyonu
Yan etkilerin raporlanması
Herhangi bir yan etki yaşarsanız, doktorunuzla veya eczacınızla konuşun.Bu kullanma talimatında listelenmeyen olası yan etkiler de buna dahildir. Yan etkileri doğrudan Ek V'te listelenen ulusal raporlama sistemi aracılığıyla da bildirebilirsiniz. Yan etkileri bildirerek bu ilacın güvenliği hakkında daha fazla bilgi sağlanmasına yardımcı olabilirsiniz.
Son Kullanma ve Saklama
Bu ilacı çocukların göremeyeceği ve erişemeyeceği bir yerde saklayın.
Bu ilacı, EXP'den sonra karton üzerinde ve EXP'den sonra her bir blisterde belirtilen son kullanma tarihinden sonra kullanmayın. Son kullanma tarihi, o ayın son gününü ifade eder.
Bu ilacı 25 °C'nin üzerinde saklamayın.
Herhangi bir ilacı atık su veya evsel atık yoluyla atmayın.Artık kullanmadığınız ilaçları nasıl atacağınızı eczacınıza sorun.Bu çevrenin korunmasına yardımcı olacaktır.
Nexavar'ın içeriği
- Aktif madde sorafenib'dir. Her film kaplı tablet 200 mg sorafenib (tosilat olarak) içerir.
- Diğer bileşenler şunlardır: Tablet çekirdeği: kroskarmeloz sodyum, mikrokristalin selüloz, hipromelloz, sodyum lauril sülfat ve magnezyum stearat. Tablet kaplama: hipromelloz, makrogol, titanyum dioksit (E 171) ve kırmızı demir oksit (E 172)
Nexavar'ın görünüşü ve paketin içeriği
Nexavar 200 mg film kaplı tabletler, bir tarafında Bayer çaprazı ve diğer tarafında "200" yazan kırmızı ve yuvarlaktır.Her biri 28 tabletlik dört şeffaf takvim blisteri içeren 112 tabletlik kartonlarda sunulurlar.
Kaynak Paket Broşürü: AIFA (İtalyan İlaç Ajansı). Ocak 2016'da yayınlanan içerik. Mevcut bilgiler güncel olmayabilir.
En güncel sürüme erişmek için AIFA (İtalyan İlaç Ajansı) web sitesine erişmeniz önerilir. Sorumluluk reddi ve faydalı bilgiler.
01.0 TIBBİ ÜRÜNÜN ADI
NEXAVAR 200 MG
02.0 KALİTATİF VE KANTİTATİF BİLEŞİM
Her film kaplı tablet 200 mg sorafenib (tosilat olarak) içerir.
Yardımcı maddelerin tam listesi için bölüm 6.1'e bakın.
03.0 FARMASÖTİK FORM
Film kaplı tablet (tablet).
Kırmızı, yuvarlak, bikonveks, bir tarafında Bayer çarpı işareti ve diğer tarafında "200" ile işaretlenmiş film kaplı tabletler.
04.0 KLİNİK BİLGİLER
04.1 Terapötik endikasyonlar
hepatokarsinom
Nexavar, hepatosellüler karsinom tedavisi için endikedir (bkz. bölüm 5.1).
böbrek hücreli karsinom
Nexavar, daha önce interferon alfa veya interlökin-2 tedavisinde başarısız olan veya bu tür bir tedavi için uygun olmadığı düşünülen ileri böbrek hücreli karsinomlu hastaların tedavisinde endikedir.
Farklılaşmış tiroid kanseri
Nexavar, lokal olarak ilerlemiş veya metastatik, ilerleyici, radyoiyodine dirençli diferansiye tiroid kanseri (papiller / foliküler / Hürthle hücresi) olan hastaların tedavisinde endikedir.
04.2 Pozoloji ve uygulama yöntemi
Nexavar ile tedavi, antikanser tedavilerinin kullanımında deneyimli bir doktorun gözetimi altında yapılmalıdır.
Dozaj
Yetişkinler için önerilen Nexavar dozu günde iki kez 400 mg sorafenib'dir (iki adet 200 mg tablet) (toplam günlük 800 mg doza eşdeğer).
Tedavi, klinik fayda gözlemlendiği sürece veya kabul edilemez toksisiteler ortaya çıkana kadar devam etmelidir.
Dozajın ayarlanması
Şüpheli advers ilaç reaksiyonlarının yönetimi, sorafenib tedavisinin geçici olarak kesilmesini veya dozunun azaltılmasını gerektirebilir.
Hepatoselüler karsinom tedavisi sırasında doz azaltılması gerektiğinde (hepatosellüler kanser, HCC) ve renal hücreli karsinom (böbrek hücreli karsinom, RCC), Nexavar dozu günde bir kez iki adet 200 mg sorafenib tablete düşürülmelidir (bkz. bölüm 4.4).
Diferansiye tiroid kanseri tedavisi sırasında doz azaltılması gerektiğinde (diferansiye tiroid karsinomu, DTC), Nexavar dozu bölünmüş dozlar halinde günde 600 mg sorafenib'e düşürülmelidir (iki 200 mg tablet ve on iki saat arayla bir 200 mg tablet).
Dozun daha da azaltılması gerekiyorsa, Nexavar bölünmüş dozlar halinde günde 400 mg sorafenib'e (on iki saat arayla iki 200 mg tablet) ve gerekirse günde bir kez 200 mg'lık bir tablete daha da düşürülebilir. advers reaksiyonlar, Nexavar dozu arttırılabilir.
Pediatrik popülasyon
Nexavar'ın daha büyük çocuklarda ve ergenlerde güvenliği ve etkinliği
Yaşlı nüfusu
Yaşlı popülasyon için (65 yaş üstü hastalar) doz ayarlaması gerekli değildir.
Böbrek yetmezliği
Hafif, orta veya şiddetli böbrek yetmezliği olan hastalarda doz ayarlaması gerekli değildir. Diyalize giren hastalar için veri mevcut değildir (bkz. bölüm 5.2).
Böbrek yetmezliği riski olan hastalarda su ve elektrolit dengesinin izlenmesi tavsiye edilir.
Karaciğer yetmezliği
Child Pugh A veya B (hafif ila orta) karaciğer yetmezliği olan hastalarda doz ayarlaması gerekli değildir. Şiddetli Child Pugh C karaciğer yetmezliği olan hastalara ilişkin veri bulunmamaktadır (bkz. bölüm 4.4 ve 5.2).
Uygulama yöntemi
Ağız yoluyla kullanım için
Sorafenib öğün aralarında veya az veya orta yağlı bir öğünle birlikte uygulanmalıdır. Hasta yağ içeriği yüksek bir yemek yemeyi planlıyorsa, sorafenib tabletleri yemekten en az bir saat önce veya iki saat sonra alınmalı ve tabletler bir bardak su ile yutulmalıdır.
04.3 Kontrendikasyonlar
Etkin maddeye veya bölüm 6.1'de listelenen yardımcı maddelerden herhangi birine karşı aşırı duyarlılık.
04.4 Özel uyarılar ve uygun kullanım önlemleri
dermatolojik toksisite
El-ayak deri reaksiyonu (palmar-plantar eritrodisestezi) e döküntü sorafenib için en yaygın advers reaksiyonları temsil eder. Döküntü ve el-ayak cilt reaksiyonu, i'ye göre genellikle Derece 1 ve 2'dir. Ortak Toksisite Kriterleri (CTC) ve genellikle sorafenib tedavisinin ilk altı haftasında ortaya çıkar. Dermatolojik toksisitenin yönetimi, semptomları hafifletmek için topikal tedavileri, tedavinin geçici olarak kesilmesini ve/veya sorafenib dozunda bir değişikliği veya şiddetli veya inatçı vakalarda uygulamasının kesin olarak kesilmesini içerebilir (bkz. bölüm 4.8).
Hipertansiyon
Sorafenib ile tedavi edilen hastalarda arteriyel hipertansiyon insidansı daha yüksek gözlenmiştir.Bu hastalarda hipertansiyon genellikle hafif ila orta derecelidir, tedavinin erken evrelerinde ortaya çıkmıştır ve standart antihipertansif tedaviye yanıt vermiştir. Kan basıncı düzenli olarak izlenmeli ve mevcut tıbbi uygulamalara göre gerektiği şekilde tedavi edilmelidir. Şiddetli veya kalıcı hipertansiyon veya hipertansif kriz durumunda, antihipertansif tedaviye başlanmasına rağmen, sorafenib uygulamasının kalıcı olarak kesilmesinin düşünülmesi önerilir (bkz. bölüm 4.8).
kanama
Sorafenib uygulamasını takiben kanama riski artabilir. Bir kanama epizodu tıbbi müdahale gerektiriyorsa, sorafenib uygulamasının kalıcı olarak kesilmesinin düşünülmesi önerilir (bkz. bölüm 4.8).
Kardiyak iskemi ve/veya kalp krizi
Çift kör, randomize, plasebo kontrollü bir çalışmada (çalışma 1, bkz. bölüm 5.1), tedavi başlangıçlı kalp enfarktüsü veya iskemi insidansı sorafenib grubunda (%4.9) tedavi grubuna göre daha yüksekti. %) Çalışma 3'te (bkz. bölüm 5.1), tedavi başlangıçlı kardiyak enfarktüs veya iskemi insidansı sorafenib ile tedavi edilen hastalarda %2.7 ve plasebo ile tedavi edilen hastalarda %1.3 olmuştur. Stabil olmayan koroner arter hastalığı veya yakın zamanda miyokard enfarktüsü geçirmiş hastalar bu çalışmaların dışında tutulmuştur. Kardiyak iskemi ve/veya enfarktüs gelişen hastalarda sorafenib tedavisinin geçici veya kalıcı olarak kesilmesi gerektiği düşünülmelidir (bkz. bölüm 4.8).
QT aralığı uzaması
Sorafenib'in QT / QTc aralığını uzattığı gösterilmiştir (bkz. Bölüm 5.1), bu da ventriküler aritmi riskinde artışa yol açabilir Konjenital Uzun QT'si olan hastalar gibi QTc uzaması olan veya gelişebilecek hastalarda sorafenib dikkatli kullanın. Sendrom, yüksek kümülatif doz antrasiklin ile tedavi edilenler, belirli antiaritmik ilaçlar veya QT uzamasına yol açabilecek diğer ilaçları alan hastalar ve hipokalemi, hipokalsemi veya hipomagnezemi gibi elektrolit bozuklukları olanlar Bu hastalarda sorafenib kullanıldığında, periyodik elektrokardiyografi tedavi süresince elektrolit (magnezyum, potasyum ve kalsiyum) ölçümleri yapılmalıdır.
Gastrointestinal perforasyon
Gastrointestinal perforasyon nadir görülen bir olaydır ve sorafenib alan hastaların %1'inden azında bildirilmiştir. Bazı durumlarda, bariz karın içi tümör ile bir ilişki yoktu. Gastrointestinal perforasyon durumunda sorafenib uygulaması kesilmelidir (bkz. bölüm 4.8).
Karaciğer yetmezliği
Şiddetli karaciğer yetmezliği (Child Pugh C) olan hastalarda veri bulunmamaktadır. Bu tür hastalarda sorafenib esas olarak karaciğer yoluyla elimine edildiğinden maruziyet artabilir (bkz. bölüm 4.2 ve 5.2).
Varfarin ile eşzamanlı uygulama
Seyrek kanama atakları veya INR'de artış (Uluslararası Normalleştirilmiş
Oran) sorafenib tedavisi sırasında varfarin alan bazı hastalarda bildirilmiştir. Varfarin veya fenprokumon tedavisi gören hastalar, protrombin zamanı, INR veya klinik olarak anlamlı kanama epizodlarındaki değişiklikler açısından düzenli olarak izlenmelidir (bkz. bölüm 4.5 ve 4.8).
Yara iyileşmesinde komplikasyonlar
Sorafenib'in yara iyileşmesi üzerindeki etkisine ilişkin resmi bir çalışma yapılmamıştır.Majör cerrahi geçiren hastalarda ihtiyati nedenlerle sorafenib tedavisinin geçici olarak durdurulması önerilir.Majör cerrahi sonrası tedaviye ne zaman yeniden başlanacağına ilişkin klinik deneyim sınırlıdır. Bu nedenle, majör cerrahiyi takiben sorafenib tedavisine devam etme kararı, yeterli yara iyileşmesinin klinik değerlendirmesine dayanmalıdır.
Yaşlı nüfusu
Böbrek yetmezliği vakaları bildirilmiştir. Bu nedenle, böbrek fonksiyonunun izlenmesi düşünülmelidir.
İlaçlar arasındaki etkileşim
Sorafenib, ağırlıklı olarak UGT1A1 (örn.
Sorafenib ve dosetakselin birlikte uygulanması durumunda dikkatli olunması önerilir (bkz. bölüm 4.5).
Neomisin veya gastrointestinal mikroflorada ciddi ekolojik bozukluklara neden olabilen diğer antibiyotiklerle kombinasyon, sorafenibin biyoyararlanımında bir azalmaya yol açabilir (bkz. bölüm 4.5) Sorafenib'in plazma konsantrasyonunda azalma riski, başlamadan önce değerlendirilmelidir. antibiyotik tedavisi kursu.
Platin bazlı kemoterapi ile kombinasyon halinde sorafenib ile tedavi edilen skuamöz hücreli akciğer kanserli hastalarda daha yüksek mortalite gözlendi.
Küçük hücreli dışı akciğer kanserli hastaları inceleyen iki randomize klinik çalışmada (Kucuk hucreli olmayan akciger kanseri, NSCLC), skuamöz hücreli akciğer kanserli bir hasta alt grubunda genel sağkalım için tehlike oranı (HR), paklitaksel / karboplatin tedavisine ek olarak sorafenib ile tedavi edilen hastalarda 1.81 (%95 GA 1.19, 2.74) ve 1.22 (%95) idi. CI 0.82; 1.80) gemsitabin / sisplatin tedavisine ek olarak sorafenib ile tedavi edilen hastalarda. Hiçbir baskın ölüm nedeni gözlenmedi, ancak platin bazlı tedaviye ek olarak sorafenib ile tedavi edilen hastalarda solunum yetmezliği, kanama ve enfeksiyon insidansında artış gözlendi.
Patolojiye özel uyarılar
Diferansiye Tiroid Karsinomu (DTC)
Tedaviye başlamadan önce, doktorlara maksimum lezyon boyutuna (bkz. bölüm 5.1), hastalıkla ilgili semptomlara (bkz. bölüm 5.1) ve ilerleme hızına göre bireysel hasta prognozunu dikkatlice değerlendirmeleri önerilir.
Şüpheli advers ilaç reaksiyonlarının yönetimi, sorafenib tedavisinin "geçici olarak kesilmesini veya doz azaltılmasını" gerektirebilir. Çalışma 5'te (bkz. bölüm 5.1), deneklerin %37'si tedaviyi geçici olarak bırakmış ve %35'i sorafenib tedavisinin 1. döngüsü kadar erken bir sürede dozu azaltmıştır.
Doz azaltma, advers reaksiyonların hafifletilmesinde yalnızca kısmen etkili olmuştur.Bu nedenle, antitümör aktivitesi ve tolere edilebilirlik dikkate alınarak tekrarlanan fayda ve risk değerlendirmeleri önerilir.
DTC'de kanama
Potansiyel kanama riski nedeniyle, DTC'li hastalara sorafenib uygulanmadan önce trakeal, bronşiyal ve özofagus infiltrasyonu lokal tedavi ile tedavi edilmelidir.
DTC'de hipokalsemi
DTC'li hastalarda sorafenib kullanırken, kan kalsiyum düzeylerinin yakından izlenmesi önerilir. Klinik çalışmalarda, böbrek hücreli karsinom veya hepatokarsinomlu hastalarla karşılaştırıldığında, DTC'li hastalarda, özellikle hipoparatiroidizm öyküsü olan hastalarda, hipokalsemi daha sık ve daha şiddetliydi. Derece 3 ve 4 hipokalsemi meydana geldi. DTC'li sorafenib ile tedavi edilen hastalar (bkz. bölüm 4.8). QT aralığı uzaması veya torsades de pointes gibi komplikasyonları önlemek için şiddetli hipokalsemi düzeltilmelidir (QT aralığının uzaması bölümüne bakınız).
DTC'de TSH'nin Bastırılması
Çalışma 5'te (bkz. bölüm 5.1), sorafenib ile tedavi edilen hastalarda TSH düzeylerinde 0,5 mU/L'den fazla artışlar gözlenmiştir. DTC'li hastalarda sorafenib kullanırken TSH düzeylerinin yakından izlenmesi önerilir.
böbrek hücreli karsinom
MSKCC prognostik grubu tarafından tanımlanan yüksek riskli hastalar (Memorial Sloan Kettering Kanser Merkezi), renal hücreli karsinomda faz III klinik çalışmasına dahil edilmemiştir (bkz. çalışma 1, bölüm 5.1) ve bu hastalarda yarar-risk oranı belirlenmemiştir.
04.5 Diğer tıbbi ürünlerle etkileşimler ve diğer etkileşim biçimleri
Metabolik enzimlerin indükleyicileri
Tek doz sorafenib uygulamasından 5 gün önce rifampisin uygulaması sorafenib EAA'sında ortalama %37 azalma ile sonuçlanmıştır Diğer CYP3A4 ve/veya glukuronidasyon indükleyicileri (örn. hipericum perforatum "St. John's wort" olarak da bilinen fenitoin, karbamazepin, fenobarbital ve deksametazon) sorafenib metabolizmasını artırabilir ve böylece konsantrasyonunu azaltabilir.
CYP3A4 inhibitörleri
Sağlıklı erkek gönüllülere 7 gün boyunca günde bir kez uygulanan güçlü bir CYP3A4 inhibitörü olan ketokonazol, 50 mg'lık tek bir sorafenib dozunun ortalama EAA değerini değiştirmemiştir.Bu veriler, sorafenib ile CYP3A4 inhibitörlerinin klinik farmakokinetik etkileşimlerinin olası olmadığını düşündürmektedir.
CYP2B6, CYP2C8 ve CYP2C9 substratları
Laboratuvar ortamında sorafenib, CYP2B6, CYP2C8 ve CYP2C9'u neredeyse eşit güçte inhibe eder. Bununla birlikte, klinik farmakokinetik çalışmalarda, günde iki kez 400 mg sorafenibin siklofosfamid, bir CYP2B6 substratı veya bir CYP2C8 substratı olan paklitaksel ile birlikte uygulanması, klinik olarak anlamlı inhibisyonla sonuçlanmamıştır.Bu veriler, sorafenib'in önerilen 400 günde iki kez mg, inhibitör olmayabilir canlıda CYP2B6 veya CYP2C8.
Ayrıca, bir CYP2C9 substratı olan sorafenib ve varfarin ile eşzamanlı tedavi, plaseboya kıyasla ortalama PT-INR'de değişikliklere yol açmamıştır. Bu nedenle, aynı zamanda bir "inhibisyon" riski canlıda sorafenib tarafından klinik olarak anlamlı CYP2C9'un düşük olduğu kabul edilebilir. Bununla birlikte, varfarin veya fenprokumon alan hastaların INR'leri düzenli olarak izlenmelidir (bkz. bölüm 4.4).
CYP3A4, CYP2D6 ve CYP2C19 substratları
Sırasıyla CYP3A4, CYP2D6 ve CYP2C19 sitokromları için substratlar olan sorafenib ve midazolam, dekstrometorfan veya omeprazolün birlikte uygulanması bu ajanlara maruziyeti değiştirmemiştir.Bu, sorafenib'in bu izoenzimlerin ne inhibitörü ne de indükleyicisi olduğunu gösterir. sitokrom P450, bu nedenle sorafenibin bu enzimlerin substratları ile klinik farmakokinetik etkileşimleri olası değildir.
UGT1A1 ve UGT1A9 substratları
Laboratuvar ortamında, sorafenib, UGT1A1 ve UGT1A9 tarafından glukuronidasyonu inhibe etti. Bu bulgunun klinik önemi bilinmemektedir (aşağıya ve bölüm 4.4'e bakınız).
Eğitim laboratuvar ortamında CYP sisteminin enzimlerinin indüksiyonu üzerine
İnsan hepatosit kültürlerinin sorafenibe maruz kalmasından sonra CYP1A2 ve CYP3A4 aktiviteleri değişmemiştir, bu da sorafenibin CYP1A2 ve CYP3A4 indükleyicisi olma olasılığının düşük olduğunu göstermektedir.
P-gp için yüzeyler
Laboratuvar ortamında, sorafenib'in taşıma proteini p-glikoproteini (P-gp) inhibe ettiği gösterilmiştir. Sorafenib ile eşzamanlı tedavi durumunda, digoksin gibi P-gp substratlarının plazma konsantrasyonunda bir artış göz ardı edilemez.
Diğer antineoplastik ajanlarla ilişki
Klinik çalışmalarda sorafenib, gemsitabin, sisplatin, oksaliplatin, paklitaksel, karboplatin, kapesitabin, doksorubisin, irinotekan, dosetaksel ve siklofosfamid dahil olmak üzere, yaygın olarak kullanılan pozolojilerinde bir dizi başka antineoplastik ajanla birlikte uygulanmıştır. Sorafenib, gemsitabin, sisplatin, karboplatin, oksaliplatin veya siklofosfamidin farmakokinetiği üzerinde klinik olarak anlamlı bir etkiye sahip değildir.
Paklitaksel / karboplatin
• Paklitaksel (225 mg/m2) ve karboplatinin (EAA = 6) sorafenib (günde iki kez ≤ 400 mg) ile birlikte uygulanması, sorafenib uygulamasına 3 gün ara verilerek (önceki iki gün ve paklitaksel/karboplatin uygulamasının yapıldığı gün) ), paklitakselin farmakokinetiği üzerinde önemli bir etkisi yoktu.
• Paklitaksel (225 mg/m2, 3 haftada bir) ve karboplatinin (EAA = 6) sorafenib (günde iki kez 400 mg, sorafenib dozlamasına ara vermeden) ile birlikte uygulanması sorafenib maruziyetinde %47'lik bir artışa, %29'luk bir artışa neden olmuştur. paklitaksel maruziyetinde artış ve 6-OH paklitaksel maruziyetinde %50 artış Karboplatinin farmakokinetiği etkilenmemiştir.
Bu veriler, paklitaksel ve karboplatin, sorafenib uygulamasına 3 gün ara verilerek (paklitaksel/karboplatin uygulamasından iki gün önce ve uygulama günü) sorafenib ile birlikte uygulandığında doz ayarlaması gerekmediğini göstermektedir. Doza ara vermeden sorafenib ile birlikte uygulanmasından hemen sonra sorafenib ve paklitaksele maruziyet artışı.
kapesitabin
Kapesitabin (günde iki kez 750-1050 mg/m2, 21 günde bir 1-14. günler) ve sorafenibin (doza ara vermeden günde iki kez 200 veya 400 mg) birlikte uygulanması, maruziyette önemli değişikliklere yol açmamış, ancak 15 Kapesitabin maruziyetinde %50 artış ve 5-FU maruziyetinde %0-52 artış 5-FU maruziyetindeki bu küçük, mütevazı artışların klinik önemi bilinmemektedir Kapesitabin ve 5-FU sorafenib ile birlikte uygulandığında.
Doksorubisin / İrinotekan
Sorafenib ile eşzamanlı tedavi, doksorubisinin EAA'sında %21'lik bir artışa neden olmuştur.Metaboliti SN-38 daha sonra UGT1A1 yolu yoluyla metabolize edilen irinotekan ile uygulandığında, "SN-38'in EAA'sında %67-120'lik bir artış olmuştur ve irinotekanın EAA'sında %26 - 42." Bu verilerin klinik önemi bilinmemektedir (bkz. bölüm 4.4).
dosetaksel
Dosetaksel (21 günde bir 75 veya 100 mg / m2'lik bir doz), sorafenib (21 günlük tedavinin 2 ila 19'uncu günlerinde günde iki kez 200 mg veya 400 mg) ile birlikte, doketaksel'e tekabül eden 3 günlük bir kesinti ile uygulanır. uygulaması) dosetaksel EAA ve Cmaks değerlerinde sırasıyla %36-80 ve %16-32 artışa neden olmuştur.Sorafenib ve dosetaksel ile birlikte uygulandığında dikkatli olunması önerilir (bkz. bölüm 4.4).
Diğer acentelerle ortaklık
neomisin
Gastrointestinal florayı yok etmek için kullanılan, sistemik olmayan bir antimikrobiyal ajan olan neomisin ile kombinasyon, sorafenibin enterohepatik devridaimini engelleyerek (bkz. bölüm 5.2, Biyotransformasyon ve Metabolizma) sorafenib maruziyetini azaltır. günlerde, ortalama sorafenib maruziyeti %54 azaldı. Diğer antibiyotiklerin etkileri araştırılmamıştır, ancak büyük olasılıkla glukuronidaz aktivitesi olan mikroorganizmalara müdahale etme yeteneklerine bağlı olacaktır.
04.6 Hamilelik ve emzirme
Gebelik
Hamile kadınlarda sorafenib kullanımına ilişkin veri yoktur Hayvan çalışmaları malformasyonlar dahil üreme toksisitesi göstermiştir (bkz. Bölüm 5.3) Sorafenib ve metabolitlerinin sıçanlarda plasentayı geçtiği gösterilmiştir ve sorafenib zararlı etkilere neden olması beklenmektedir. Sorafenib, açıkça gerekli olmadıkça ve sadece annenin ihtiyaçları ve fetüs üzerindeki riskler dikkatle değerlendirildikten sonra hamilelik sırasında kullanılmamalıdır.
Çocuk doğurma potansiyeli olan kadınlar tedavi sırasında etkili doğum kontrolü kullanmalıdır.
Besleme zamanı
Sorafenib'in insan sütüne geçip geçmediği bilinmemektedir. Hayvanlarda sorafenib ve/veya metabolitleri süte geçer Sorafenib yenidoğanın büyümesini ve gelişmesini bozabileceğinden (bkz. Bölüm 5.3), kadınlar sorafenib tedavisi sırasında emzirmeyi bırakmalıdır.
Doğurganlık
Hayvan çalışmaları, sorafenib'in erkek ve kadın doğurganlığını bozabileceğini göstermektedir (bkz. bölüm 5.3).
04.7 Araç ve makine kullanma yeteneği üzerindeki etkiler
Araç veya makine kullanma yeteneği üzerine herhangi bir çalışma yapılmamıştır. Sorafenib'in araç veya makine kullanma yeteneğini etkilediğine inanmak için hiçbir neden yoktur.
04.8 İstenmeyen etkiler
En önemli ciddi advers reaksiyonlar miyokardiyal iskemi ve enfarktüs, gastrointestinal perforasyon, ilaç hepatiti, kanama ve hipertansiyon veya hipertansif krizdi.
En yaygın advers reaksiyonlar diyare, asteni, alopesi, enfeksiyon, el-ayak deri reaksiyonu (MedDRA'da "palmar-plantar eritrodisestezi sendromuna" karşılık gelir) ve döküntü.
Farklı klinik çalışmalarda veya pazarlama sonrası kullanımda bildirilen advers reaksiyonlar MedDRA'ya ve sıklığa göre sıralanmış olarak tablo 1'de listelenmiştir Sıklıklar şu şekilde tanımlanmıştır: çok yaygın (≥1/10 ), yaygın (≥1/100,
Her bir sıklık sınıfı içinde, istenmeyen etkiler azalan önem sırasına göre sunulmaktadır.
Tablo 1: Farklı klinik çalışmalarda veya pazarlama sonrası kullanımda hastalarda bildirilen genel advers reaksiyonlar.
* Advers reaksiyonlar yaşamı tehdit edici veya ölümcül olabilir. Bu olaylar ya nadirdir ya da yaygın olmayandan daha az sıklıkta görülür.
** El-ayak cilt reaksiyonu, MedDRA'daki palmar-plantar eritrodisestezi sendromuna karşılık gelir
Bazı advers reaksiyonlar hakkında daha fazla bilgi edinin
Konjestif kalp yetmezliği
şirket sponsorluğundaki bir klinik çalışmada, sorafenib ile tedavi edilen hastaların %1.9'unda (N = 2276) konjestif kalp yetmezliği advers olay olarak rapor edilmiştir. Çalışma 11213'te (RCC) konjestif kalp yetmezliği ile uyumlu advers olaylar sorafenib ile tedavi edilen hastaların %1.7'sinde ve plasebo ile tedavi edilen hastaların %0.7'sinde bildirilmiştir. Çalışma 100554'te (HCC) bu tür olaylar sorafenib ile tedavi edilen hastaların %0.99'unda ve plasebo ile tedavi edilen hastaların %1.1'inde rapor edilmiştir.
Özel popülasyonlar için ek bilgiler
Klinik çalışmalarda, el-ayak deri reaksiyonu, ishal, alopesi, kilo kaybı, hipertansiyon, hipokalsemi ve keratoakantom / skuamöz hücreli deri karsinomu gibi bazı advers ilaç reaksiyonları, diferansiye tiroid kanserli hastalarda hastalara kıyasla oldukça yüksek bir sıklıkta meydana geldi. renal veya hepatoselüler hücreli karsinom çalışmalarına dahil edilmiştir.
HCC (çalışma 3) ve RCC (çalışma 1) hastalarında laboratuvar testlerindeki değişiklikler
Lipaz ve amilazda bir artış çok yaygın olarak bildirilmiştir Lipazda Derece 3 veya 4 artış Ortak Toksisite KriterleriAdvers Olaylar (CTCAE), Çalışma 1 (RCC) ve Çalışma 3'te (HCC) sorafenib grubundaki hastaların sırasıyla %11 ve %9'unda meydana gelirken, Sorafenib grubundaki hastaların %7 ve %9'unda meydana gelmiştir. Çalışma 1 ve çalışma 3'teki sorafenib grubundaki hastaların sırasıyla %1 ve %2'sinde, her iki plasebo grubundaki hastaların %3'ünde bir CTCAE Derece 3 veya 4 amilaz yükselmesi meydana geldi.451 hastanın 2'sinde klinik pankreatit bildirildi Çalışma 1'de sorafenib (CTCAE Derece 4), Çalışma 3'te sorafenib (CTCAE Derece 2) ile tedavi edilen 297 hastanın 1'i ve çalışma 1'de plasebo ile tedavi edilen 451 hastadan (CTCAE Derece 2) 1'i (CTCAE Derece 2) ile tedavi edilmiştir.
Hipofosfatemi çok yaygın bir laboratuvar bulgusudur ve çalışma 1 ve çalışma 3'te sorafenib ile tedavi edilen hastaların sırasıyla %45 ve %35'inde, plasebo ile tedavi edilen hastaların sırasıyla %12 ve %11'inde gözlenmiştir. CTCAE Derece 3 hipofosfatemi (1 - 2 mg / dL) çalışma 1'de sorafenib ile tedavi edilen hastaların %13'ünde ve plasebo ile tedavi edilen hastaların %3'ünde meydana gelirken, çalışma 3'te sorafenib ile tedavi edilen hastaların %11'inde ve Plasebo ile tedavi edilen hastaların %2'si Hiçbir CTCAE Derece 4 hipofosfatemi vakası bildirilmemiştir (sorafenib ile ilişkili hipofosfateminin etiyolojisi bilinmemektedir.
Sorafenib ile tedavi edilen hastaların ≥ %5'inde lenfopeni ve nötropeni dahil CTCAE derece 3 veya 4 laboratuvar anormallikleri gözlenmiştir.
Çalışma 1 ve çalışma 3'te plasebo grubundaki hastaların sırasıyla %7,5 ve %14.8'ine kıyasla sorafenib ile tedavi edilen hastaların %12 ve %26.5'inde hipokalsemi gözlenmiştir. Hipokalsemi vakaları hafif olmuştur (CTCAE derece 1 ve 2). CTCAE derece 3 hipokalsemi (6.0 - 7.0 mg/dL), sorafenib ile tedavi edilen hastaların %1.1 ve %1.8'inde ve plasebo grubundaki hastaların %0.2 ve %1.1'inde meydana geldi ve bir CTCAE derece 4 hipokalsemi (
Çalışma 1 ve 3'te, plasebo alan hastaların %0,7 ve %5,9'una kıyasla sorafenib ile tedavi edilen hastaların sırasıyla %5.4 ve %9.5'inde potasyumda bir azalma gözlemlenmiştir. Çoğu hipokalemi vakası hafifti (CTCAE derece 1). Bu çalışmalarda, sorafenib ile tedavi edilen hastaların %1,1 ve %0,4'ünde ve plasebo grubundaki hastaların %0,2 ve %0,7'sinde CTCAE derece 3 hipokalemi meydana gelmiştir. CTCAE derece 4 hipokalemi vakaları.
DTC'li hastalarda laboratuvar testlerindeki değişiklikler (çalışma 5)
Plasebo grubundaki hastaların %11.0'ına kıyasla sorafenib ile tedavi edilen hastaların %35.7'sinde hipokalsemi gözlenmiştir. Hipokalsemi vakalarının çoğu hafif şiddetteydi. CTCAE derece 3 hipokalsemi sorafenib ile tedavi edilen hastaların %6,8'inde ve plasebo grubundaki hastaların %1.9'unda meydana gelirken, CTCAE derece 4 hipokalsemi sorafenib ile tedavi edilen hastaların %3.4'ünde ve plasebo grubundaki hastaların %1.0'ında meydana geldi.
Çalışma 5'te gözlemlenen diğer klinik olarak ilgili laboratuvar değişiklikleri Tablo 2'de gösterilmiştir.
Tablo 2: Çift kör fazda DTC hastalarında (çalışma 5) bildirilen tedaviyle ortaya çıkan laboratuvar anormallikleri
* Advers Olaylar için Ortak Terminoloji Kriterleri (CTCAE), sürüm 3.0
** Sorafenib ile ilişkili hipofosfateminin etiyolojisi bilinmemektedir.
Şüpheli advers reaksiyonların raporlanması
Tıbbi ürünün yarar/risk oranının sürekli olarak izlenmesine olanak sağladığından, tıbbi ürünün ruhsatlandırılmasından sonra meydana gelen şüpheli advers reaksiyonların raporlanması önemlidir.Sağlık profesyonellerinden, İtalyan İlaç Kurumu, web sitesi aracılığıyla herhangi bir şüpheli advers reaksiyonu bildirmeleri istenmektedir. : www.agenziafarmaco.gov.it/it/responsabili.
04.9 Doz aşımı
Sorafenib doz aşımı durumunda spesifik bir tedavi yoktur. Klinik olarak incelenen en yüksek sorafenib dozu günde iki kez 800 mg'dır. Bu dozajı takiben gözlenen yan etkiler başlıca diyare ve dermatolojik reaksiyonlardır. Doz aşımından şüpheleniliyorsa sorafenib kesilmeli ve gerekirse destekleyici tedavi başlatılmalıdır.
05.0 FARMAKOLOJİK ÖZELLİKLER
05.1 Farmakodinamik özellikler
Farmakoterapötik grup: antineoplastik ajanlar, protein kinaz inhibitörleri.
ATC kodu: L01XE05.
Sorafenib, hem anti-proliferatif hem de anti-anjiyojenik özellikler gösteren bir kinaz inhibitörüdür. laboratuvar ortamında ve canlıda.
Etki mekanizması ve farmakodinamik etkiler
Sorafenib, kanser hücrelerinin çoğalmasını engelleyen bir kinaz inhibitörüdür. laboratuvar ortamında. Sorafenib, atimik farelere nakledilen geniş bir insan tümör spektrumunun büyümesini engelleyerek tümör anjiyogenezinde bir azalmaya neden olur Sorafenib, tümör hücresinde bulunan hedeflerin (CRAF, BRAF, V600E BRAF, c-KIT ve FLT-) aktivitesini inhibe eder. 3) ve tümörün kan damarlarında (CRAF, VEGFR-2, VEGFR-3 ve PDGFR-ß). RAF kinazlar serin/treonin kinazlar iken c-KIT, FLT-3, VEGFR-2, VEGFR-3 ve PDGFR-ü reseptör tirozin kinazlardır.
Klinik etkinlik
Sorafenib'in güvenliliği ve klinik etkinliği hepatoselüler karsinomalı hastalarda incelenmiştir (hepatosellüler kanser, HCC), ilerlemiş renal hücreli karsinomalı hastalarda (böbrek hücreli karsinom, RCC) ve diferansiye tiroid kanserli hastalarda (diferansiye tiroid karsinomu, DTC).
hepatokarsinom
Çalışma 3 (çalışma 100554) hepatoselüler kanserli 602 hastada çok merkezli, randomize, çift kör, plasebo kontrollü, uluslararası bir Faz III klinik çalışmasıydı. Temel demografik ve hastalık özellikleri, Estern Kooperatif Onkoloji Grubu (ECOG) sınıflandırmasına göre sorafenib ve plasebo grupları arasında karşılaştırılabilirdi (derece 0: %54'e karşı %54; derece 1: %38'e karşı %39; derece 2: %8'e karşı %8 %7), TNM sınıflandırmasına (aşama I:
Çalışma, planlanmış bir geçici Genel Hayatta Kalma (OS) analizinin önceden tanımlanmış etkinlik sınırını aşmasının ardından kapatıldı. Bu OS analizi, plasebo ile tedavi edilen hastalara kıyasla sorafenib ile tedavi edilen hastalarda OS'de istatistiksel olarak anlamlı bir artış gösterdi (HR: 0.69, p = 0.00058, bkz. tablo 3).
Bu çalışmada Child Pugh B karaciğer bozukluğu olan hastalardaki veriler sınırlıdır ve yalnızca bir Child Pugh C hastası dahil edilmiştir.
Tablo 3: Hepatokarsinomda Çalışma 3'ten (Çalışma 100554) Etkililik Sonuçları
CI = Güven aralığı, HR = Tehlike oranı (plaseboya göre sorafenib)
* p değeri, 0,0077 olarak ayarlanan varsayılan O "cutoff sınırının altında olduğu için istatistiksel olarak anlamlıdır
** bağımsız radyolojik inceleme
İkinci bir Faz III, uluslararası, çok merkezli, randomize, çift kör, plasebo kontrollü çalışma (Çalışma 4, 11849), ilerlemiş karaciğer kanseri olan 226 hastada sorafenibin klinik yararını değerlendirdi. Çin, Kore ve Tayvan'da yürütülen bu çalışma, sorafenib'in olumlu fayda-risk profili (HR (OS): 0.68, p = 0.01414) ile ilgili olarak Çalışma 3'ün sonuçlarını doğrulamıştır.
Çalışma 3 ve 4'ün önceden tanımlanmış katmanlaştırma faktörlerinde (ECOG sınıflandırması, makroskopik vasküler invazyon varlığı veya yokluğu ve/veya tümörün ekstrahepatik yayılımı) HR tutarlı bir şekilde plaseboya göre sorafenib lehine olmuştur. Keşif amaçlı alt grup analizleri, zaten başlangıçta uzak metastazları olan hastalarda daha az belirgin bir tedavi etkisi önerdi.
böbrek hücreli karsinom
İleri böbrek hücreli karsinom (RCC) tedavisinde sorafenib'in tolere edilebilirliği ve etkinliği iki klinik çalışmada incelenmiştir:
Çalışma 1 (çalışma 11213), 903 hastada çok merkezli, randomize, çift kör, plasebo kontrollü bir Faz III klinik çalışmaydı. Yalnızca berrak hücreli böbrek tümörü olan ve MSKCC'ye göre düşük ve orta risk faktörü olan hastalar alındı. NS uç noktabirincil genel sağkalım idi (OS, etraflı hayatta kalma) ve progresyonsuz sağkalım (PFS, ilerleme Özgür hayatta kalma).
Hastaların yaklaşık yarısının ECOG skalasına göre genel durumu 0'dı ve hastaların yarısı MSKCC sınıflamasına göre düşük skorlu prognostik gruba aitti.
PFS, kör bir bağımsız radyolojik inceleme ile RECIST kriterlerine göre değerlendirildi. 769 hastada 342 olay üzerinde PFS analizi yapılmıştır.Plasebo alan hastalarda 84 gün ile karşılaştırıldığında sorafenib ile tedavi edilen hastalarda medyan PFS değeri 167 gün olmuştur (HR = 0.44; %95 GA : 0.35 - 0.55; p
Bir analiz geçici (ikinci analiz geçici) genel sağkalım için (etraflı hayatta kalma) 903 hastada 367 ölüm üzerinde yapılmıştır. Bu analiz için nominal alfa değeri 0,0094'tür. Medyan sağkalım, sorafenib ile tedavi edilen hastalarda 19.3 ay iken, plaseboya randomize edilen hastalarda 15.9 aydı (HR = 0.77; %95 GA: 0.63-0.95; p = 0.015). Analiz sırasında yaklaşık 200 hasta plasebo grubundan sorafenib grubuna geçmiştir.
Çalışma 2, RCC de dahil olmak üzere metastatik kanserli hastalarda tedavinin randomize olarak sonlandırıldığı bir Faz II çalışmasıydı. Stabil hastalığı olan ve sorafenib tedavisi alan hastalar, plaseboya veya sorafenib tedavisine devam edilmek üzere randomize edildi. RCC'li hastalarda PFS anlamlı olarak daha yüksekti (163 gün) sorafenib ile tedavi edilen hastalarda, plasebo alan hastalarda gözlenenden daha fazla (41 gün) (p = 0.0001, HR = 0.29).
Diferansiye tiroid karsinomu (DTC)
Çalışma 5 (çalışma 14295), lokal olarak ilerlemiş veya metastatik radyoiyodine dirençli DTC'si olan 417 hastada yürütülen uluslararası, çok merkezli, randomize, çift kör, plasebo kontrollü bir faz III çalışmasıydı. RECIST kriterlerine dayalı kör bağımsız radyolojik değerlendirme ile belirlenen progresyonsuz sağkalım (PFS), çalışmanın birincil son noktasıydı.İkincil sonlanım noktaları genel sağkalım (OS), tümör yanıt oranı ve yanıt süresini içeriyordu. açık etiketli sorafenib alın.
Kayıttan önceki 14 ay içinde ilerleme gösteren ve radyoiyodine dirençli bir DTC'si olan hastalar çalışmaya dahil edildi (radyoaktif iyot, RAI). RAI'ye dirençli DTC, RAI sintigrafisinde iyot arttırıcı olmayan bir lezyonun varlığı veya kümülatif RAI ≥ 22,2 GBq uygulaması veya önceki 16 ay içinde RAI tedavisinden sonra progresyon olarak tanımlandı. birbirinden maksimum 16 ay mesafe.
Temel demografik özellikler ve hasta özellikleri, iki tedavi grubunda iyi dengelenmiştir. Hastaların %86'sında akciğerlerde, %51'inde lenf düğümlerinde ve %27'sinde kemikte metastaz mevcuttu. Kayıttan önce uygulanan medyan kümülatif radyoiyodin aktivitesi yaklaşık 14.8 GBq idi. Çoğu hastada papiller karsinom (%56.8) vardı, bunu foliküler karsinom (%25.4) ve kötü diferansiye karsinom (%9.6) izliyordu.
PFS'ye kadar geçen medyan süre, plasebo grubunda 5,8 aya kıyasla sorafenib grubunda 10.8 aydı. (HR = 0,587; %95 güven aralığı (GA): 0,454, 0,758; p tek taraflı
Sorafenib'in PFS üzerindeki etkisi, coğrafi bölge, 60 yaş üstü veya altı, cinsiyet, histoloji ve kemik metastazlarının varlığı veya yokluğundan bağımsız olarak sabitti.
Nihai PFS analizi için kesim tarihinden 9 ay sonra yapılan genel bir sağkalım analizinde, tedavi grupları arasında genel sağkalımda istatistiksel olarak anlamlı bir fark yoktu (HR 0.884; CI %95: 0.633; 1.236, tek taraflı p- 0.236 değeri). Plasebo kolunda 36.5 ay iken ortanca OS elde edilmedi.
Çift kör fazda medyan tedavi süresi sorafenib alan hastalar için 46 hafta (aralık 0.3-135) ve plasebo alan hastalar için 28 haftadır (aralık 1.7-132).
Tam bir yanıt gözlenmedi (tam yanıt, CR) RECIST kriterlerine göre. Genel yanıt oranı (CR + kısmi yanıt, kısmi yanıt (PR)), sorafenib grubunda (24 hasta, %12,2) plasebo grubuna (1 hasta, %0,5) kıyasla daha yüksekti, p tek taraflı
Maksimal tümör boyutuna dayalı alt grupların post-post analizi, maksimal tümör lezyonu boyutu 1,5 cm veya daha fazla olan hastalarda plaseboya kıyasla PFS üzerinde sorafenib lehine bir tedavi etkisi gösterdi (HR 0,54 (%95 GA: 0.41-0.71)) maksimum tümör lezyonu boyutu 1.5 cm'den küçük olan hastalarda sayısal olarak daha düşük bir etki kaydedilirken (HR 0.87 (%95 GA: 0.40 -1.89)).
Başlangıç koşullarında mevcut olan tiroid kanserine bağlı semptomlara dayalı bir post-hoc analiz, hem semptomatik hem de asemptomatik hastalarda plaseboya göre sorafenib lehine PFS üzerinde bir tedavi etkisi gösterdi Serbest sağkalım ilerlemesi için HR değeri 0.39 (%95 GA: 0.21 - Başlangıçta semptomları olan hastalar için 0.72) ve bazal koşullarda semptomu olmayan hastalar için 0.60 (%95 GA: 0.45 - 0.81).
QT aralığının uzaması
Bir klinik farmakoloji çalışmasında, başlangıçta (tedavi öncesi) ve tedaviden sonra 31 hastada QT/QTc ölçülmüştür. 28 günlük bir tedavi döngüsünden sonra, maksimum sorafenib konsantrasyonu zamanında, plasebo grubu için başlangıca kıyasla QTcB 4 ± 19 msn ve QTcF 9 ± 18 msn uzamıştır. Tedavi sonrası EKG izleme sırasında hiçbir hastada QTcB veya QTcF değeri > 500 msn göstermedi (bkz. bölüm 4.4).
Pediatrik popülasyon
Avrupa İlaç Ajansı, böbrek ve renal pelvis kanseri için pediatrik popülasyonun tüm alt gruplarında (nefroblastom, nefroblastomatozis, berrak hücreli sarkom, mezoblastik nefrom, renal medüller karsinom ve böbreğin rabdoid tümörü hariç) çalışmaların sonuçlarını sunma zorunluluğundan feragat etmiştir. ve karaciğer ve intrahepatik safra kanalı karsinomu (hepatoblastom hariç) ve diferansiye tiroid karsinomu (pediatrik kullanım hakkında bilgi için bkz. bölüm 4.2).
05.2 "Farmakokinetik özellikler
Emilim ve dağıtım
Sorafenib tabletlerinin uygulanmasından sonra, oral bir çözelti ile karşılaştırıldığında ortalama nispi biyoyararlanım %38 - 49'dur. Mutlak biyoyararlanım bilinmemektedir. Oral uygulamayı takiben sorafenib, yaklaşık 3 saatte doruk plazma seviyelerine ulaşır. Yağ oranı yüksek bir yemekle birlikte verildiğinde, sorafenib emilimi, aç durumda uygulamaya kıyasla yaklaşık %30 oranında azalır.
Ortalama Cmaks ve EAA, günde iki kez 400 mg'ın üzerindeki dozlarla orantılı olarak daha az artar. Sorafenib'in plazma proteinlerine bağlanması laboratuvar ortamında %99,5'tir.
Sorafenib'in 7 gün boyunca tekrarlanan dozları, tek uygulamaya kıyasla 2.5 ila 7 kat birikme ile sonuçlanmıştır. Sorafenib'in kararlı durumuna 7 gün içinde ulaşılır ve ortalama zirve/düşük plazma konsantrasyonları oranı 2'den azdır.
DTC, RCC ve HCC hastalarında günde iki kez 400 mg uygulanan sorafenib denge konsantrasyonları belirlendi En yüksek ortalama konsantrasyon DTC'li hastalarda gözlendi (RCC ve HCC hastalarında gözlenenin yaklaşık iki katı), ancak değişkenlik yüksekti. tüm tümör tipleri DTC hastalarındaki bu yüksek konsantrasyonun nedeni bilinmemektedir.
Biyotransformasyon ve eliminasyon
Sorafenib'in eliminasyon yarı ömrü yaklaşık 25 ila 48 saattir. Sorafenib, esas olarak karaciğerde CYP3A4 aracılı oksidatif metabolizma ve UGT1A9 aracılı glukurono-konjugasyon yoluyla metabolize edilir. Konjuge sorafenib, bazı bakterilerin glukuronidaz aktivitesi ile gastrointestinal kanalda salınabilir, böylece konjuge olmayan aktif bileşenin yeniden emilmesine izin verir Neomisin ile kombinasyonun bu sürece müdahale ettiği ve sorafenib'in ortalama biyoyararlanımını %54 oranında azalttığı gözlemlenmiştir.
Sorafenib, kararlı durum plazmasında dolaşan analitlerin yaklaşık %70-85'ini oluşturur. Beşi plazmada bulunan sekiz sorafenib metaboliti tanımlanmıştır. Plazmada dolaşan sorafenib'in ana metaboliti olan piridin N-oksit, potens sergiler.laboratuvar ortamında sorafenibe benzer. Bu metabolit, kararlı durumda dolaşan analitlerin yaklaşık %9 - 16'sını oluşturur.
100 mg'lık bir sorafenib çözeltisinin oral yoldan verilmesini takiben, dozun %96'sı 14 gün içinde geri kazanılmıştır: %77'si feçeste ve %19'u idrarda glukuronat metabolitleri olarak. Dozun %51'ini temsil eden değişmemiş sorafenib, dışkıda geri kazanılmış, ancak idrarda bulunmamıştır; bu, metabolize edilmemiş aktif maddenin safra yoluyla atılımının sorafenibin eliminasyonuna katkıda bulunabileceğini göstermektedir.
Belirli hasta kategorilerinde farmakokinetik
Demografik verilerin analizi, farmakokinetik ile yaş (65 yaşına kadar), cinsiyet veya vücut ağırlığı arasında bir ilişki olmadığını göstermiştir.
Pediatrik popülasyon
Pediyatrik hastalarda sorafenib farmakokinetiğini doğrulamak için hiçbir çalışma yapılmamıştır.
Yarış
Beyaz ve Asyalı denekler arasında farmakokinetik açısından klinik olarak anlamlı farklılıklar yoktur.
Böbrek yetmezliği
Dört Faz I klinik çalışmada, hafif veya orta derecede böbrek yetmezliği olan hastalarda kararlı durum sorafenib maruziyeti, normal böbrek fonksiyonu olan hastalarda bulunana benzer olmuştur.Klinik bir farmakoloji çalışmasında (400 mg tek doz sorafenib), Normal böbrek fonksiyonu veya hafif, orta veya şiddetli böbrek yetmezliği olan hastalarda sorafenib maruziyeti ve böbrek fonksiyonu. Diyaliz gerektiren hastalarda veri bulunmamaktadır.
Karaciğer yetmezliği
Hepatosellüler karsinomlu hastalarda (HCC) ve Child-Pugh A veya B (hafif ila orta) olarak değerlendirilen karaciğer yetmezliği ile maruziyet değerleri karşılaştırılabilirdi ve karaciğer yetmezliği olmayan hastalarda gözlenen aralık içindeydi. HCC'si olmayan Child-Pugh A ve B hastalarında sorafenib farmakokinetiği, sağlıklı gönüllülerde görülene benzerdi. Şiddetli (Child-Pugh C) karaciğer yetmezliği olan hastalara ilişkin veri bulunmamaktadır. Sorafenib esas olarak karaciğer yoluyla elimine edilir ve bu hasta popülasyonunda maruziyet artabilir.
05.3 Klinik öncesi güvenlik verileri
Sorafenib'in klinik öncesi güvenlik profili fareler, sıçanlar, köpekler ve tavşanlarda değerlendirilmiştir.
Tekrarlanan doz toksisite çalışmaları, klinik çalışmalarda kullanılan dozların altındaki maruziyetlerde (EAA karşılaştırmasına dayalı olarak) çeşitli organlarda (dejenerasyon ve rejenerasyon) değişiklikleri ortaya çıkarmıştır.
Genç ve büyümekte olan köpeklerde tekrarlanan dozlamadan sonra, klinik çalışmalarda kullanılan dozların altındaki maruziyetlerde kemikler ve dişler üzerinde etkiler gözlenmiştir. Bu etkiler, femurun büyüme plakasının düzensiz kalınlaşması, değişen büyüme plakalarının çevresinde medüller hipoplazi ve dentin bileşimindeki değişikliklerden oluşuyordu. Yetişkin köpekte benzer etkiler indüklenmedi.
Standart genotoksisite çalışmaları programı yürütüldü ve bir tahlilde kromozomal yapısal anormalliklerde bir artış kaydedildiği için pozitif sonuçlar elde edildi. laboratuvar ortamında Metabolik aktivasyon varlığında klastojenisitenin ölçümü için memeli hücrelerinde (Çin hamsteri yumurtalıkları). Sorafenib, Ames testinde veya mikronükleus testinde genotoksik değildi canlıda farede. Nihai aktif maddede de bulunan üretim sürecinin bir ara maddesi (bakteri hücreleri üzerinde in vitro (Ames testi).Ek olarak, standart genotoksik pilde test edilen sorafenib partisi, %0.34 PAPE içeriyordu.
Sorafenib ile karsinojenisite çalışmaları yapılmamıştır.
Fertilite üzerindeki etkisini değerlendirmek için sorafenib ile spesifik hayvan çalışmaları yapılmamıştır. Bununla birlikte, tekrarlanan doz hayvan çalışmaları, klinik deneylerde (EAA'ya dayalı olarak) kullanılan dozajların altındaki maruziyetlerde erkek ve dişi üreme organlarında değişiklikler gösterdiğinden, erkek ve dişi doğurganlığı üzerinde olumsuz bir etki beklenebilir. sıçanlarda testis, epididim, prostat ve seminal veziküllerin dejenerasyon belirtileri ve gecikmiş gelişimi Dişi sıçanlar korpus luteinin merkezi nekrozu ve yumurtalıklarda foliküler gelişmenin tıkanması gösterdi Köpekler yumurtalıklarda tübüler dejenerasyon, testisler ve oligospermi gösterdi.
Sorafenib, klinik çalışmalarda kullanılan dozların altındaki maruziyetlerde sıçanlara ve tavşanlara uygulandığında embriyotoksik ve teratojenik olduğu gösterilmiştir. Gözlenen etkiler arasında maternal ve fetal vücut ağırlığında azalma, fetal rezorpsiyon sayısında artış ve eksternal ve viseral malformasyon sayısında artış yer aldı.
Çevresel risk değerlendirme çalışmaları, sorafenib tosilatın potansiyel olarak kalıcı, biyolojik birikim yapan ve çevre için toksik olduğunu göstermiştir.Çevresel risk değerlendirmesine ilişkin bilgiler bu tıbbi ürün için Avrupa Kamu Değerlendirme Raporunda (EPAR) mevcuttur (bakınız bölüm 6.6 ).
06.0 FARMASÖTİK BİLGİLER
06.1 Yardımcı maddeler
tabletin çekirdeği:
kroskarmeloz sodyum
Mikrokristal selüloz
hipromelloz
Sodyum lauril sülfat
Magnezyum stearat
Tablet kaplama:
hipromelloz
makrogol
Titanyum dioksit (E 171)
Kırmızı demir oksit (E 172)
06.2 Uyumsuzluk
İlgili değil.
06.3 Geçerlilik süresi
3 yıl.
06.4 Depolama için özel önlemler
25 °C'nin üzerinde saklamayın.
06.5 İç ambalajın yapısı ve paketin içeriği
Şeffaf blisterde (PP / Alüminyum) 112 film kaplı tablet (4 x 28) içeren karton.
06.6 Kullanım ve kullanım talimatları
Bu tıbbi ürün çevre için potansiyel bir risk oluşturabilir.Kullanılmayan tıbbi ürün ve bu tıbbi üründen elde edilen atıklar yerel yönetmeliklere uygun olarak atılmalıdır.
07.0 PAZARLAMA YETKİ SAHİBİ
Bayer İlaç AG
13342 Berlin
Almanya
08.0 PAZARLAMA YETKİ NUMARASI
AB / 1/06/342/001
037154010
09.0 İLK İZİN VEYA İZİNİN YENİLENMESİ TARİHİ
İlk izin tarihi: 19 Temmuz 2006
En son yenileme tarihi: 21 Temmuz 2011
10.0 METİN REVİZYON TARİHİ
05/2014











-e-ruscogenina.jpg)