genellik
Osteogenezis imperfekta, cinsiyetle bağlantılı olmayan, belirli bir kemik kırılganlığından ve belirgin bir kırılma eğiliminden sorumlu olan doğuştan genetik bir hastalıktır.
Osteogenezis imperfekta semptomları çoktur; genellikle, kemik zayıflaması, kemik kırılmalarına yüksek eğilim, mavi, gri veya mor oküler sklera varlığı, kemik deformitelerinin veya diğer iskelet değişikliklerinin varlığı, üçgen yüz, diş kırılganlığı vb. .
Genel olarak, osteogenezis imperfekta'nın doğru teşhisi için aşağıdakiler gereklidir: fizik muayene, tıbbi öykü, tıbbi görüntüleme testleri, tip I kollajen değerlendirme testi ve genetik test.
Ne yazık ki, şu anda osteogenezis imperfektalı hastalar için mevcut olan tek tedavi semptomatiktir. Söz konusu hastalık aslında tedavi edilemez.
Osteogenezis imperfekta nedir?
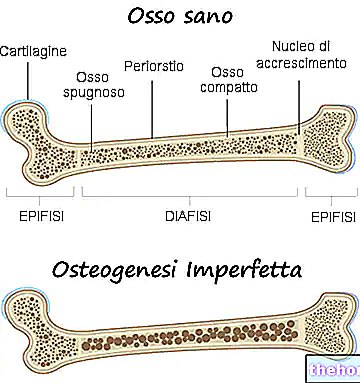
Osteogenezis imperfekta, etkilenen kişinin kemiklerini zayıflatan ve kırılmaya daha yatkın hale getiren genetik bir hastalıktır.
Gerçekte, osteogenezis imperfekta terimi ile doktorlar, belirli bir derecede kemik kırılganlığı ile karakterize edilen heterojen bir genetik hastalık grubuna atıfta bulunur. Bu nedenle, bazıları diğerlerinden çok daha şiddetli olan çeşitli osteogenezis imperfekta formları (veya türleri) vardır.
DOĞUMSAL BİR HASTALIKTIR
Bundan etkilenen kişilerde osteogenezis imperfekta doğuştan gelen bir hastalıktır ve bu nedenle tüm niyet ve amaçlarla doğuştan gelen bir hastalık olarak tanımlanabilir.
SEKS İLE İLGİLİ Mİ?
Osteogenezis imperfekta, hemofili veya Klinefelter sendromu gibi cinsiyete bağlı genetik bir hastalık değildir.
EPİDEMİLOJİ
Bazı istatistiksel araştırmalara göre, osteogenezis imperfekta insidansı her 15.000-20.000 doğumda bir vakaya eşit olacaktır. Bu, her 15.000-20.000 yeni doğan bebeğin osteogenezis imperfektadan etkilendiği anlamına gelir.
Diğer istatistiksel çalışmalar, osteogenezis imperfekta'nın erkekleri ve kadınları eşit olarak etkilediğini ve belirli bir popülasyon veya etnik grup için herhangi bir tercihi olmadığını göstermiştir.
Yaşam süresi, osteogenezis imperfekta formuna bağlı olan son derece değişken bir parametredir.
nedenler
Osteogenezis imperfekta hemen hemen her zaman tip I kollajen üretiminin kalitatif ve kantitatif değişiminden kaynaklanır.
Tip I kollajen, kemikleri güçlendirmek ve kıkırdak, tendonlar, deri, oküler sklera vb. oluşturan sağlıklı bağ dokularını korumak için gereklidir.
Bu nedenle, tip I kollajen üretimindeki bir değişiklik, kemiklerin gücünü ve insan vücudunda bulunan bağ dokularının sağlığını etkiler.
KOLAJEN ÜRETİMİNİ NE DEĞİŞTİRİR?
Genetik bir hastalık, hücresel DNA'yı oluşturan bir veya daha fazla genin mutasyona uğraması nedeniyle ortaya çıkan bir durumdur.
Osteogenezis imperfekta durumunda, ikincisinin nedenleri hemen hemen her zaman COL1A1 (kromozom 17'de bulunur) ve COL1A2 (kromozom 7'de bulunur) genlerinden birinin veya her ikisinin mutasyonunda bulunur.
Normal koşullar altında, COL1A1 ve COL1A2, tip I kollajenin normal üretimini düzenler; sorumluluklarında mutasyonların varlığında, düzenleyici işlevlerinde başarısız olurlar.
Önemli: Mutasyona uğrarsa başka hangi genler osteogenezis imperfektaya neden olur?
COL1A1 ve COL1A2 mutasyonlarına ek olarak, IFITM5, SERPINF1, CRTAP ve LEPRE1 genlerindeki mutasyonlar osteogenezis imperfekta'nın potansiyel nedenleridir.
Yukarıda bahsedilen genler, COL1A1 ve COL1A2'den farklı işlevleri kapsar - bu nedenle tip I kolajen üretimini kontrol etmezler - ancak yine de "insan iskeletinin kemiklerinin gücü ve direnci üzerinde bir etkiye sahiptirler.
NE TÜR GENETİK HASTALIKTIR?
Osteogenezis imperfekta otozomal genetik bir hastalıktır.
Genetik bir hastalıkla ilişkili otozomal terimi, söz konusu durumun otozomal ve cinsiyet dışı kromozomlara dayalı genetik mutasyonlardan kaynaklandığını belirtir.
Okuyuculara, insanın 22 çift otozomal tipte ve sadece bir çift cinsel tip olmak üzere toplam 23 çift kromozomdan oluşan bir kromozom setine sahip olduğu hatırlatılır. bireysel.
COL1A1, COL1A2 ve IFITM5 mutasyonlarını takiben oluşan osteogenezis imperfekta, otozomal dominant bir hastalığın tüm özelliklerini taşır.SERPINF1, CRTAP ve LEPRE1 genlerindeki mutasyonlara bağlı olduğunda, otozomal resesif hastalık özelliklerine sahiptir.
TÜRLER
Şu anda doktorlar, 8 tip (veya form) osteogenezis imperfekta olduğuna inanmaktadır. Çeşitli türleri ayırt etmek için, ilk sekiz Romen rakamını kesin olarak Roma numaralandırmasını kullanmaya karar verdiler.
Aşağıdaki tablo, osteogenezis imperfekta'nın 8 formunu, bunlara neden olan mutasyonları ve diğer özellikleri göstermektedir.
Adam
mutasyona uğramış gen
Genetik hastalık türü
NS
COL1A1
otozomal dominant
II
COL1A1 ve COL1A2
otozomal dominant
III
COL1A1 ve COL1A2
otozomal dominant
IV
COL1A1 ve COL1A2
otozomal dominant
V.
IFITM5
otozomal dominant
SEN
SERPINF1
otozomal çekinik
VII
CRTAP
otozomal çekinik
VIII
tavşan 1
otozomal çekinik
* Not: Açıkçası, osteogenezis imperfekta'nın ilk dört formuna neden olan COL1A1 ve COL1A2'deki mutasyonlar, biraz farklı özelliklere sahip genetik değişikliklerdir. Aksi takdirde, birini diğerinden ayırt etmenin bir anlamı olmayacaktır.
Belirtiler, belirtiler ve komplikasyonlar
Tüm osteogenezis imperfekta türleri, kemiklerin zayıflamasından sorumludur, öyle ki hastalıktan etkilenen kişinin kırılmaya karşı özel bir eğilimi vardır. Kemiklerin zayıflama derecesi şekle göre değişir; bunlardan bazıları için bu zayıflama diğerlerinden daha fazladır.
Bunu söyledikten sonra, her osteogenezis imperfekta formunun, bazıları için diğer formların semptomatolojik resmini hatırlatabilecek kendi semptomatik tablosuna sahip olduğuna işaret edilmelidir.
OLASI BELİRTİLER VE İŞARETLER
Osteogenezis imperfekta'nın olası semptomları ve belirtileri şunları içerir:
- Kemik malformasyonlarının varlığı;
- Kısa ve küçük bir gövdenin varlığı (gövde olarak tasarlanmıştır);
- Eklem sorunları (ör: gevşek eklemler);
- Kas Güçsüzlüğü;
- Mavi, mor veya gri oküler sklera;
- Üçgen yüz;
- varil göğüs;
- Omurga kolonunun morfolojik anomalileri;
- Diş kırılganlığı;
- İşitme kaybı veya tamamen kaybı;
- Solunum Problemleri
- Tip 1 kolajenin yokluğu veya yokluğu ile ilgili problemler.
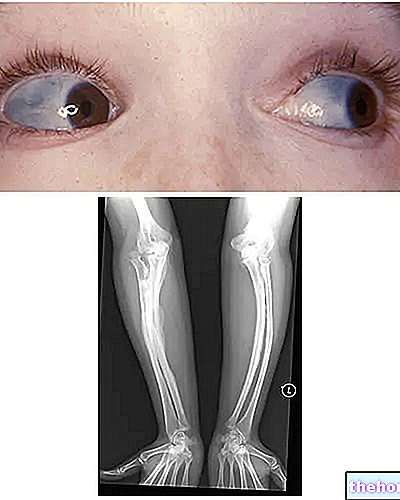
Osteogenez İmperfekta: skleraların mavi rengine ve hastalığı karakterize eden kemik deformasyonlarına dikkat edin. wikipedia.org'dan
KUSURSUZ OSTEOJENEZİN EN CİDDİ ŞEKİLLERİ NELERDİR?
Doktorlar, çeşitli osteogenezis imperfekta tiplerinin semptomatolojik şiddetini 3 derecelik bir ölçekte sınıflandırır: hafif derece, orta derece ve şiddetli derece.
Sadece bir form "hafif derece" kategorisine aittir: "tip I osteogenezis imperfekta"; 4 form osteogenezis imperfekta "orta derece" kategorisine aittir: IV, V ve VI; son olarak, "şiddetli derece" kategorisine ait 3 formlar: II, III, VII ve VIII.
TİP I: ÖZELLİKLER
En yaygın ve en az şiddetli olan tip I osteogenezis imperfekta aşağıdaki özelliklere sahiptir:
- Özellikle ergenlik öncesi kırıklara neden olur;
- Boy üzerinde neredeyse hiç etkisi yoktur, bu nedenle hastalar genellikle normal boydadır;
- Eklem sorunlarına ve kas güçsüzlüğüne neden olur
- Mavi, mor veya gri skleradan sorumludur;
- Üçgen yüz ve omurga anomalilerinin nedenidir;
- Neredeyse hiçbir zaman kemik deformitelerine neden olmaz. Onları kışkırtırsa, asgari düzeydedirler;
- Diş kırılganlığına ve / veya işitme kaybına neden olabilir (ikincisi genellikle yetişkinlikte görülür);
- Kalitesi normal ancak miktarı anormal olan tip I kollajenin varlığı ile ilişkilidir (normalden daha zayıftır).
TİP II: ÖZELLİKLER
Tip II osteogenezis imperfekta ile karakterize edilir:
- Doğumda veya kısa süre sonra ölüm nedeni. Solunum sorunları neredeyse her zaman ölüme neden olur;
- Önemli kemik kırılganlığının ve ciddi kemik deformitelerinin varlığı;
- Kısa boy ve az gelişmiş akciğerler
- Mavi, mor veya gri renkli sklera;
- Tip I kollajenin nicel ve nitel anomalilerinin varlığı.
TİP III: ÖZELLİKLER
Tip III osteogenezis imperfekta aşağıdaki özelliklere sahiptir:
- Çok ciddi olmasına rağmen yenidoğan döneminde sıklıkla ölüme neden olmaz;
- "Yüksek kemik kırılganlığı;
- Boy kısalığı, eklem sorunları, kas güçsüzlüğü (özellikle bacaklarda ve kollarda), fıçı göğüs, üçgen yüz ve omurganın anormal eğriliğinden sorumludur;
- Mavi, mor veya gri skleradan kaynaklanır;
- Solunum problemlerine, diş kırılganlığına ve işitme kaybına neden olabilir;
- Kemik deformitelerinden sıklıkla sorumludur;
- Tip I kollajenin kalitatif ve kantitatif anormallikleri ile ilişkilidir.
TİP IV: ÖZELLİKLER
Tip IV osteogenez ile karakterize edilir:
- Form II ve III ile form I arasında bir derece kemik kırılganlığı;
- Ortalama boydan daha kısa;
- Mavi, mor veya gri renkli sklera;
- Hafif / orta derecede kemik deformiteleri, omurga ve namlu göğsünde hafif anormallikler;
- Üçgen yüz;
- Diş kırılganlığının ve işitme kaybının olası varlığı;
- Tip I kollajen anormalliklerinin varlığı.
TİP V: ÖZELLİKLER
Tip V osteogenezis imperfekta bazı yönlerden tip IV osteogenez imperfektaya benzer. Ancak, bazı özellikleri vardır, bunlar:
- Normal renkli sklera;
- Diş kırılganlığının olmaması;
- Kırık kemiklerin iyileşme sürecinde anormal kemik nasırlarının oluşumu;
- Radius ve ulna arasında bulunan interosseöz membranın kalsifikasyonu. Bu, önkolun hareketliliğini bozar.
TİP VI: ÖZELLİKLER
Tip VI osteogenezis imperfekta da form IV'e benzerdir. İkincisinden ayırt etmek için, yüksek kan alkalin fosfataz seviyeleri ve bazı kemiklerde balık dikenlerine benzer lamellerin (kemikli) varlığı da dahil olmak üzere bazı özellikler vardır.
TİP VII: ÖZELLİKLER
Semptomatik olarak, tip VII osteogenezis imperfekta bazı durumlarda tip IV'e ve diğer durumlarda tip II'ye benzeyebilir.
Bu ciddi patolojik formun özellikleri şunları içerir:
- Kısa boy;
- Aşırı kısa bir humerus (kol kemiği) ve femur (uyluk kemiği) varlığı;
- Coxa vara olarak bilinen kalça deformitesinin sık görülmesi.
TİP VIII: ÖZELLİKLER
Tip VIII osteogenezis imperfekta, II ve III formlarını çok andırır.
Kendine özgü özellikleri arasında öne çıkanlar şunlardır: şiddetli büyüme eksikliği, şiddetli iskelet hipomineralizasyonu ve prolil 3-hidroksilaz enziminin yokluğu (veya çok az varlığı).
Teşhis
Genel olarak, osteogenezis imperfektadan şüphelenilen hastaların maruz kaldığı tanı süreci, dikkatli bir fizik muayene ve dikkatli bir tıbbi öykü ile başlar; daha sonra hastanın aile öyküsünün analizi ve bir dizi tanısal görüntüleme testi (X-ışınları, BT taramaları, vb.) ile devam eder; son olarak, tip I kollajenin nicel ve nitel bir değerlendirmesi ve genetik test.
Bugün, hamile bir kadını ultrasona tabi tutarak, doğum öncesi aşamada bile osteogenezis imperfekta tanısı koyma olasılığı vardır.
OBJEKTİF İNCELEMENİN ÖNEMİ VE TARİHÇESİ
Osteogenezis imperfekta konusunda uzman bir doktor, çoğu zaman, yukarıda bahsedilen hastalığı yalnızca fizik muayene ve anamnez yoluyla bile teşhis edebilir. Bu, bu teşhis testlerinin ihmal edilebilir bir öneme sahip olmadığı anlamına gelir.
TİP I KOLAJEN ÜRETİMİNİN DEĞERLENDİRİLMESİ
Kural olarak, tip I kollajenin kalitatif ve kantitatif değerlendirmesi çok güvenilir bir testtir, çünkü belirtildiği gibi, osteogenezis imperfekta vakalarının çoğu, tip 1 kollajen üretimini kontrol eden genlerdeki mutasyonlarla karakterize edilir.
Bir bireyde hücresel düzeyde bulunan tip I kolajenin miktarını ve kalitesini değerlendirmek için doktorlar cilt biyopsisine veya belirli bir kan testine güvenebilir.
Bu değerlendirme testlerinin her ikisi de oldukça karmaşıktır ve hastanın (veya ebeveynlerinin) sonuçları bilmesi için birkaç hafta beklemesi gerekebilir.
GENETİK TEST
Doktorlar, incelenen bireyin tüm DNA'sını araştıran bir genetik test sayesinde, mevcut genetik mutasyonun özelliklerini kesin olarak belirleyebilirler.
Genel olarak, tip I kollajenin özelliklerinin değerlendirilmesi istenen sonuçları vermediğinde veya "osteogenesis imperfecta"ya neden olan COL1A1 veya COL1A2'de bir mutasyon olmadığında, tüm hücresel DNA üzerinde bir genetik test yapılması öngörülmektedir.
DOĞUM ÖNCESİ TANI
Prenatal ultrason, tip II ve tip III osteogenezis imperfektayı belirlemede çok faydalıdır.
terapi
Şu anda osteogenezis imperfekta için spesifik bir tedavi yoktur.Başka bir deyişle, osteogenezis imperfekta hastaları, genellikle hastalığın kendi sonuçlarından kaynaklanan ölüme kadar yukarıda belirtilen durumla yaşamaya mahkumdur.
Spesifik tedavinin olmaması, diğer tedavi biçimlerinin varlığını dışlamaz. Aslında osteogenezis imperfektalı bir hastanın tedavi olanakları arasında çeşitli semptomatik tedaviler yer almaktadır; semptomatik tedaviler ile semptomları hafifleten, hastalığın seyrini yavaşlatan ve en ciddi sonuçları önleyen (veya en azından erteleyen) tedavileri kastediyoruz.
MUHTEMEL SEMPTOMİK TEDAVİLER
Osteogenezis imperfekta için olası semptomatik tedaviler listesinde aşağıdakiler öne çıkmaktadır:
- Kırıklara ve deformasyonlara karşı daha fazla direnç sağlayan tırnakların en uzun kemiklerin (NO: kırılmaya en yatkın olan) içine cerrahi olarak yerleştirilmesi. Bu işlem denir çubuk çekme intramedüller;
- Kırıkların ve/veya kemik deformitelerinin konservatif veya cerrahi tedavisi;
- Diş bakımı, diş sağlığını korumak için;
- Çok ağrılı çoklu kırıklarda ağrı giderici tedaviler;
- Kas uzatma ve güçlendirme için fizyoterapi Elastik ve tonik bir kas aparatı, çeşitli kemik kırıklarına yol açabilecek düşmeleri önlemenizi sağlar;
- Tekerlekli sandalyeler, diş telleri, koltuk değnekleri vb. dahil olmak üzere hareket için yardımcı araçların kullanımı.
HAREKETİN FAYDALARI
Osteogenezis imperfektalı bireyler için doktorlar, bu aktivitelerin her ikisi de iskelet ve kas sistemlerinin güçlendirilmesine katkıda bulunduğundan, genel olarak sürekli fiziksel egzersiz ve hareket uygulamasını tavsiye eder.
Önerilen sporlar arasında "iskelet sistemi üzerinde düşük etkili bir fiziksel aktivite" olduğu için yüzme ve yürüyüş yer almaktadır.
SAĞLIKLI YAŞAM TARZININ FAYDALARI
Sağlıklı bir yaşam sürmek, sigara içmekten kaçınmak, aşırı alkol tüketmek, çok fazla ve kötü yemek yemek vb., hastalığın ilerlemesini yavaşlattığı ve kemik kırılganlığını azalttığı için osteogenezis imperfektalı hastalar için ayrı sağlık yararlarından daha fazlasına sahiptir.
DENEY DÖNEMİNDE BELİRTİLEN TEDAVİLER
Şu anda doktorlar ve araştırmacılar, büyüme hormonu tedavisi ve bifosfonat bazlı intravenöz ve oral tedavi dahil olmak üzere bazı semptomatik tedavilerin etkinliğini değerlendiriyorlar.
Şu anda, yukarıda bahsedilen araştırma tedavilerinin sağladığı sonuçlar, tüm tıp camiası için iyiye işaret ediyor.
prognoz
Osteogenezis imperfekta tedavi edilemez olduğu için olumsuz prognoza sahip bir hastalıktır, yaşam kalitesini büyük ölçüde etkiler ve bazı durumlarda etkilenen kişinin erken ölümüne neden olur.
Bununla birlikte, modern semptomatik tedaviler sayesinde, hafif bir osteogenezis imperfekta formuna sahip birçok kişinin keyifli ve tatmin edici bir yaşam sürdürebildiği belirtilmelidir.
Önleme
Ne yazık ki, şu anda osteogenezis imperfektaya karşı önleyici bir önlem bulunmamaktadır.

-cos-cause-e-terapia.jpg)